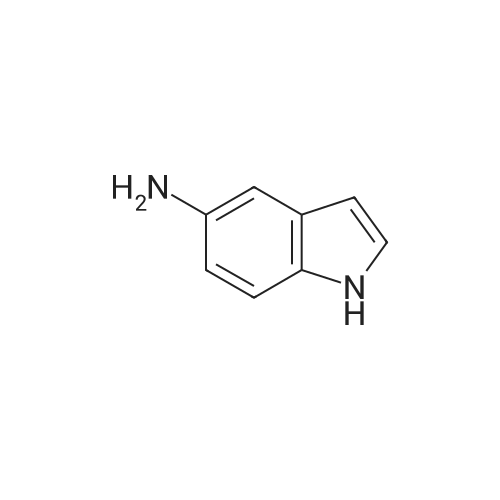
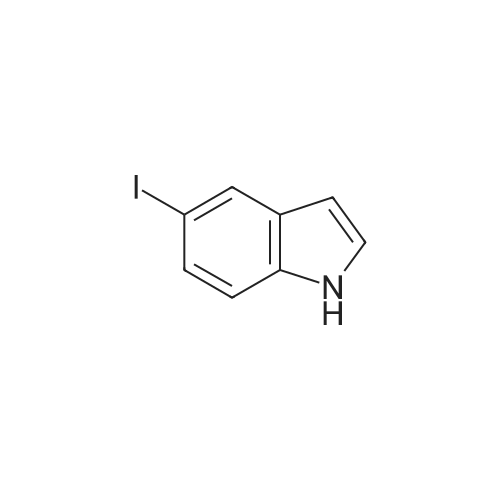
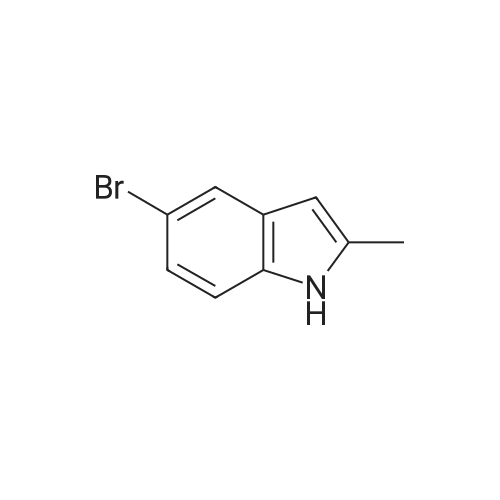
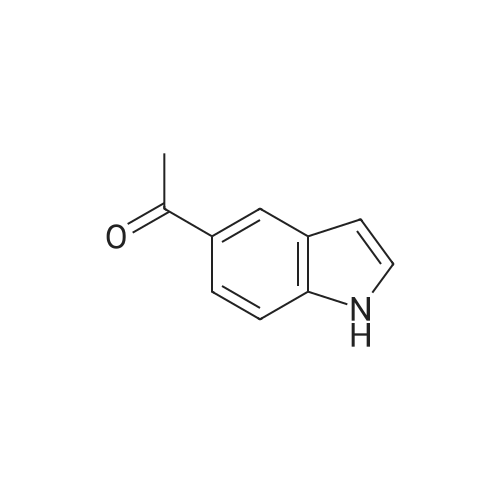
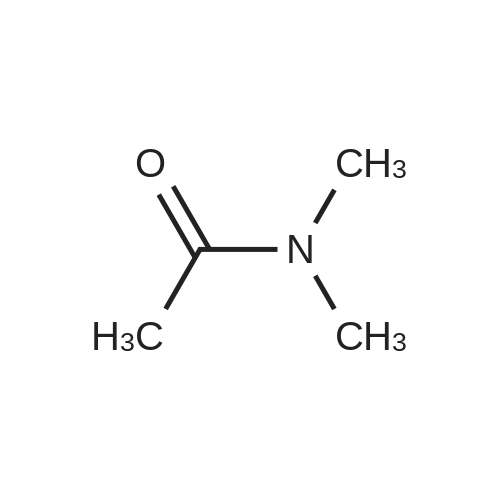
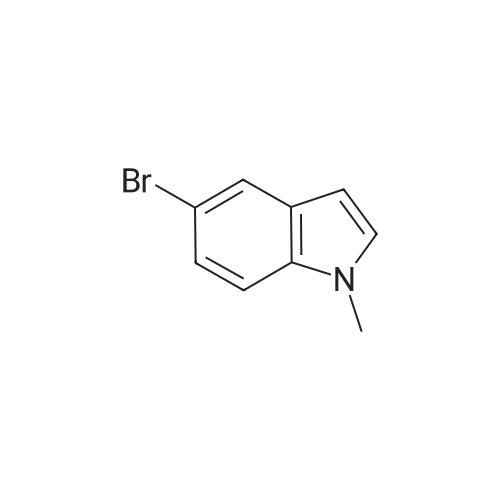
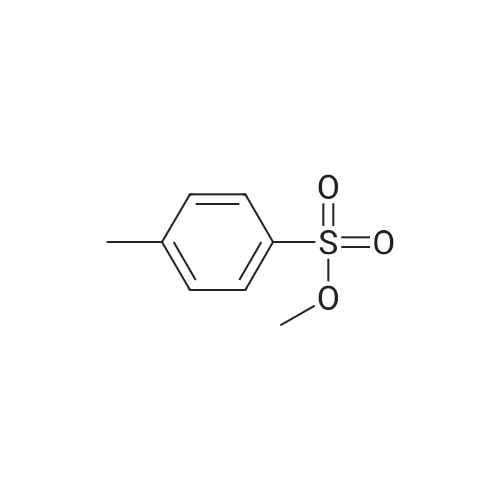
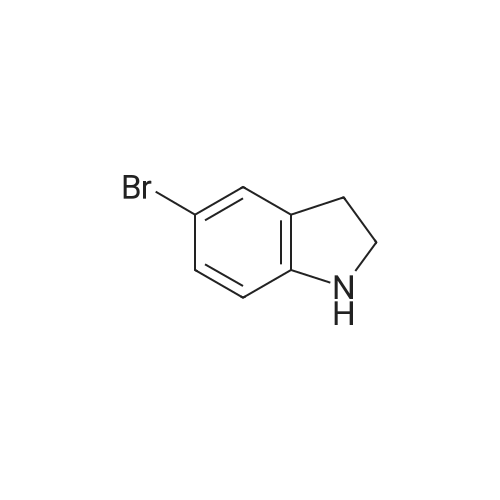
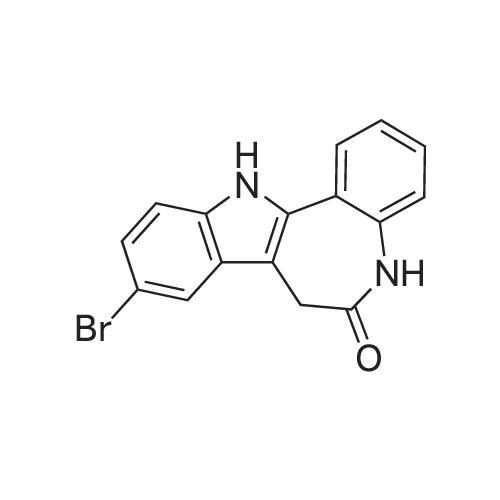
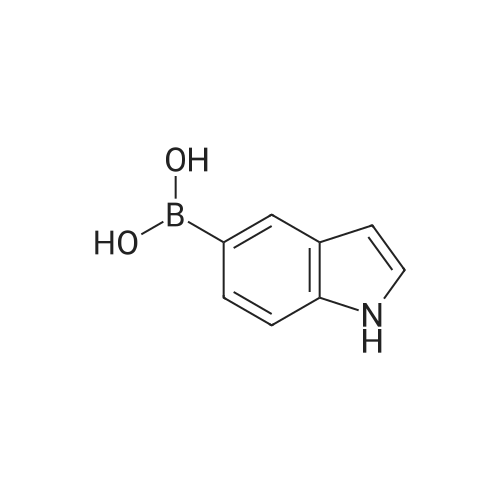
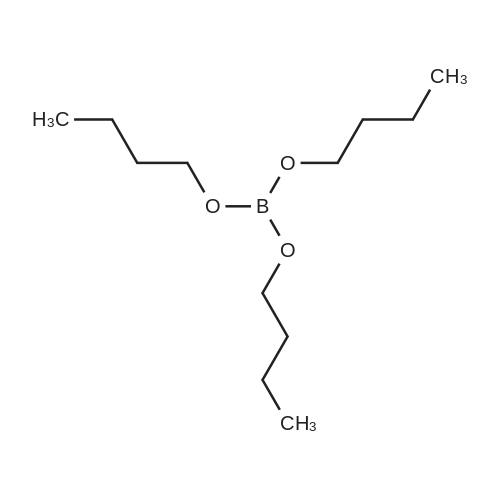
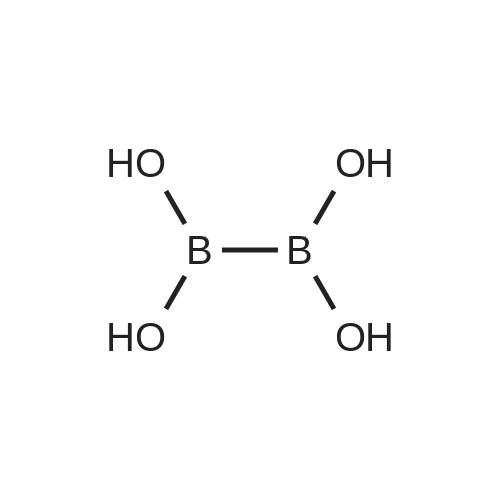
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With ammonium hydroxide; copper(I) iodide; 1-ethylacetoacetate-3-methyl imidazolium hydroxide In acetonitrile at 80℃; for 12 h; Inert atmosphere
General procedure: An oven-dried flask was charged with aryl halide (1.0 mmol), aqueous NH3 (28percent, 1.5 mmol), CuI nanoparticles (0.02 mmol), 4a (3.0 mmol) and acetonitrile (2 mL). The contents were stirred under argon atmosphere at rt for 12 h. After completion of the reaction as monitored by TLC, the product was extracted with diethyl ether (5×5 mL). The organic layer was washed with brine, dried over MgSO4 and concentrated in vacuo. Purification was done on silica gel column, and elution with ethyl acetate–hexane mixture afforded the aminated products. All products obtained herein are known compounds, and were confirmed by 1H NMR, 13C NMR and mass spectroscopic analysis, see Supplementary data for full details.
With trans-N,N'-dimethyl-1,2-cyclohexyldiamine; sodium iodide In 1,4-dioxane at 110℃; for 22 - 23 h;
A Schlenk tube was charged with CuI (9.6 mg, 0.0504 mmol, 5.0 molpercent), aryl bromide (if it is a solid; 1.00 mmol), NaI (300 mg, 2.00 mmol), briefly evacuated and backfilled with argon. [TRANS-N, NAPOS;-DIMETHYL-1,] 2-cyclohexanediamine [(16GEL,] 0. [10 MMOL,] 10 molpercent), aryl bromide (if it is a liquid; 1.00 mmol), and dioxane (1.0 mL) were added under argon. The Schlenk tube was sealed with a Teflon valve and the reaction mixture was stirred at [110 °C] for 22-23 h. The resulting suspension was allowed to reach room temperature, diluted with 30percent aq ammonia (5 [ML),] poured into water (20 mL), and extracted with dichloromethane [(3X15] mL). The combined organic phases were dried [(MGS04] or [NA2S04),] concentrated, and the residue was purified by flash chromatography on silica gel to provide the desired product.
82%
With copper(I) oxide; <i>L</i>-proline; potassium iodide In ethanol at 110℃; for 30 h; Schlenk technique; Inert atmosphere; Sealed tube
General procedure: A Schlenk tube was charged with Cu2O (7.2 mg, 10 molpercent), l-proline (11.5 mg, 20 molpercent), aryl (or heteroaryl) bromide (1 or 3,0.50 mmol), potassium iodide (KI) (249 mg, 0.75 mmol), and EtOH(1.5 mL) under nitrogen atmosphere. The Schlenk tube was sealedwith a teflon valve, and then the reaction mixture was stirred at110C for a period (the reaction progress was monitored by GCanalysis). After the reaction was completed, GC yield of high volatileproduct was determined using an appropriate internal standard(chlorobenzene or 1-chloro-4-methylbenzene) or the solvent wasremoved under reduced pressure. The residue obtained was puri-fied via silica gel chromatography (eluent: petroleum ether/ethylacetate = 10/1) to afford aryl iodides 2a–2o or heteroaryl iodides4a–4g.
Reference:
[1] Journal of the American Chemical Society, 2002, vol. 124, # 50, p. 14844 - 14845
[2] Patent: WO2004/13094, 2004, A2, . Location in patent: Page 42-43
[3] Angewandte Chemie - International Edition, 2015, vol. 54, # 1, p. 263 - 266[4] Angew. Chem., 2015, vol. 127, # 01, p. 265 - 268,4
[5] Catalysis Today, 2016, vol. 274, p. 129 - 132
[6] Chemical Communications, 2012, vol. 48, # 33, p. 3993 - 3995
[7] Journal of the American Chemical Society, 2015, vol. 137, # 26, p. 8328 - 8331
[8] Synlett, 2001, # 2, p. 266 - 268
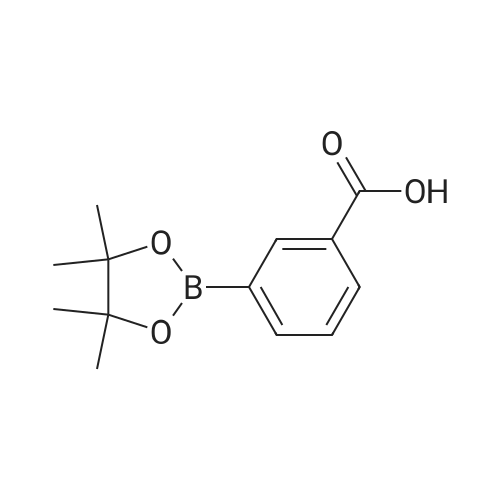
3
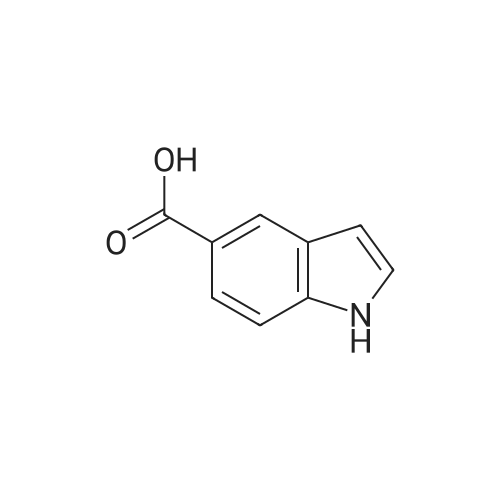
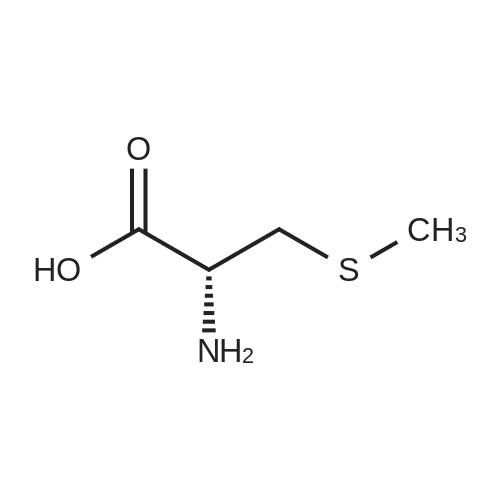
[ 10075-50-0 ]
[ 74-83-9 ]
[ 1075-34-9 ]
Reference:
[1] Journal of the American Chemical Society, 2016, vol. 138, # 35, p. 11299 - 11305
Stage #1: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5 h; Stage #2: at 20℃; for 0.166667 h;
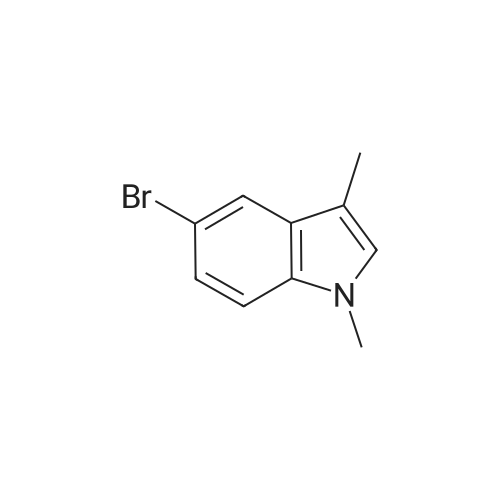
2. To solution of 5-bromo-lH-indole (2.08 g, 10.6 mmol) in 35 mL of DMF, was added sodium hydride (60 wtpercent dispersion, 467 mg, 11.67 mmol) at 0 °C. After 30 min, iodomethane(663 μL·, 10.6 mmol) was added. The reaction mixture was allowed warm to RT, and stirred atRT for 10 min. The reaction was quenched with water at 0 °C, and extracted with EtOAc (x3).The combined organic extracts were dried (MgS04), filtered, and concentrated. Flash chromatography (10 percent EtOAc/Hexanes) gave 5-bromo-l-methyl-lH-indole (2.16 g, 97percent).JHNMR (400 MHz, CDC13) δ ppm: 7.74 (d, J=1.6 Hz, IH), 7.30 (dd, J=8.4, 2.0 Hz, IH), 7.19 (d,J=8.8 Hz, IH), 7.05 (d, J=3.2 Hz IH), 6.42 (d, J=2.8 Hz, IH), 3.78 (s, 3H).
96%
Stage #1: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0 - 20℃; for 0.5 h; Stage #2: at 0 - 20℃;
General procedure: Indoles were methylated by a similar procedure to that reported in the literature.26 To a stirred suspension of NaH (60percent dispersion inmineral oil; 480 mg, 12.0 mmol) in DMF (10 mL) was added dropwise a solution of an indole (10.0 mmol) in DMF (10 mL) at 0 °C, and the mixture was stirred at room temperature for 30 min. To the mixture was added dropwise iodomethane (d 2.28; 934 mL, 15.0 mmol) at 0 °C, and the resulting mixture was stirred at room temperature overnight. The reaction was quenched with water and the aqueous layer was extracted with ethyl acetate. The combined organic layer was dried over MgSO4 and evaporated to leave a residue, which was purified by column chromatography using the indicated eluent.
95%
Stage #1: With potassium hydroxide In dimethyl sulfoxide for 1 h; Cooling with ice Stage #2: at 20℃; for 0.75 h;
Potassium hydroxide (5.74 g, 102 mmol) was dissolved in dimethylsulfoxide (85 mL) and cooled in an ice bath. 5-Bromoindole (5.00 g, 26 mmol) was added portionwise and the reaction mixture stirred for 1 h before the dropwise addition of iodomethane (7.22 g, 51 mmol). After 45 min the solution was diluted with water (85 mL). The aqueous layer was extracted with diethyl ether (3 x 50 mL). The organic layer was then washed with water (2 x 50 mL), brine (2 x 50 mL) and dried (MgSO4). The solvent was removed under reduced pressure to afford a yellow oil that was purified using column chromatography eluting with light petroleum/ethyl acetate (10:1). Yellow solid (5.20 g, 95percent). 1H NMR (400MHz, CDCl3) δ7.74 (t, 1 H, J = 2 Hz), 7.30-7.17 (m, 2 H), 7.04 (d, 1 H, J = 3 Hz), 6.41 (dd, J = 1 & 3 Hz), 3.76 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 135.4, 130.1, 130.0, 124.3, 123.3, 112.7, 110.7, 100.5, 33.0.
95.29%
Stage #1: With sodium hydride In tetrahydrofuran at 0℃; for 0.25 h; Stage #2: at 0 - 20℃; for 0.75 h;
To a stirred slurry solution of NaH (1.54 g, 1.2 eq) in THE (10 mL) at 0CC, 5-bromo- 1H-indole (5.0 g, 1.0 eq) in THE (10 mL) was added drop wise and stirred for about15 mm. Then, 0H31 (2.4 ml, 1.2 eq) was added drop wise and the resulting mixture was stirred at same temperature for about 15 mm, slowly raising the temperature of the mixture to room temperature. The mixture is continued to be stirred for another 30 mm. The reaction mixture was quenched with ice water and extracted with ethyl acetate, the combined organic layer and washed with 1 N HCI solution and water,dried and concentrated to yield the title product (5.1 g, 95.29percent) as a brownish oily mass.
91.5%
Stage #1: With sodium hydride In N,N-dimethyl-formamide at 0 - 20℃; for 0.5 h; Inert atmosphere Stage #2: at 20℃; for 1 h;
To a solution of compound 5-1 (5 g, 25.5 mmol) in dry DMF (100 mL) was added sodium hydride (NaH) (1.53 g, 38.26 mmol) under nitrogen atmosphere in an ice bath (0° C.), and then the reaction solution was warmed to r.t. and was reacted for 0.5 h. To this solution, all MeI (7.24 g, 38.26 mmol) was added at once and then the resulting solution was reacted at r.t. for 1 h. The reaction was monitored by TLC until the completion of reaction. Then saturated NH4Cl (aq.) was added to the solution to quench the reaction. The reaction solution was extracted with EtOAc (100 mL) twice, and the organic phase was washed with saturated brine (50 mL×2), dried over anhydrous Na2SO4, filtered and concentrated to remove solvent, therefore obtaining compound 5-2 (4.9 g, Yield 91.5percent). 1HNMR (400 MHz, CDCl3): δ ppm 7.74-7.75 (m, 1H), 7.28-7.31 (m, 1H), 7.18-7.21 (m, 1H), 7.04-7.06 (m, 1H), 6.41-6.43 (m, 1H), 3.78 (s, 3H)
77%
Stage #1: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5 h; Inert atmosphere Stage #2: at 20℃; for 5 h; Inert atmosphere
In a dry flask under argon, 5-bromoindole (ab) (1.5 g, 7.65 mmol) was dissolved in dry DMF (15 mL). After cooling this mixture to 0 °C, sodium hydride (367 mg, 9.18 mmol, 60percent in oil) was added to form the anion. This solution was stirred at 0 °C for 30 minutes. Then, methyl iodide (0.525 mL, 1.196 g, 8.42 mmol) was added. The resulting mixture was then stirred at room temperature for 5 hours and quenched with water. After dilution in ethyl acetate and separation, the organic layer was washed with brine three times, dried over anhydrous MgSO4 and concentrated. The desired product (ia) (1.24 g, 5.9 mmol) was obtained as a yellow solid. Yield: 77percent. 1H NMR (300 MHz, CDCl3): δ = 3.79 (s, 3H, CH3), 6.42 (d, J = 3.3 Hz, 1H, CH), 7.03 (d, J = 3.3 Hz, 1H, CH), 7.18 (d, J= 8.7 Hz, 1H, CH), 7.25-7.31 (m, 1H, CH), 7.74 (d, J= 1.8 Hz, 1H, CH) ppm.
77%
Stage #1: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5 h; Inert atmosphere Stage #2: at 20℃; for 5 h; Inert atmosphere
DMF (15 mL). After cooling this mixture to 0 °C, sodium hydride (367 mg, 9.18 mmol, 60percent in oil) was added to form the anion. This solution was stirred at 0 °C for 30 minutes. Then, methyl iodide (0.525 mL, 1.196 g, 8.42 mmol) was added. The resulting mixture was then stirred at room temperature for 5 hours and quenched with water. After dilution in ethyl acetate and separation, the organic layer was washed with brine three times, dried over anhydrous MgS04 and concentrated. The desired product (ia) (1.24 g, 5.9 mmol) was obtained as a yellow solid. Yield: 77percent.1H NMR (300 MHz, CDC13): δ = 3.79 (s, 3H, CH3), 6.42 (d, J= 3.3 Hz, 1H, CH), 7.03 (d, J= 3.3 Hz, 1H, CH), 7.18 (d, J = 8.7 Hz, 1H, CH), 7.25-7.31 (m, 1H, CH), 7.74 (d, J = 1.8 Hz, 1H, CH) ppm.
Reference:
[1] Chemistry Letters, 1991, # 7, p. 1125 - 1128
[2] Bioorganic and Medicinal Chemistry Letters, 1999, vol. 9, # 3, p. 333 - 336
[3] Organic Letters, 2015, vol. 17, # 2, p. 254 - 257
[4] Patent: WO2016/114816, 2016, A1, . Location in patent: Paragraph 0332
[5] Bioorganic and Medicinal Chemistry, 2010, vol. 18, # 12, p. 4524 - 4529
[6] Tetrahedron, 2016, vol. 72, # 5, p. 734 - 745
[7] Tetrahedron Letters, 2013, vol. 54, # 8, p. 843 - 846
[8] Patent: WO2014/202580, 2014, A1, . Location in patent: Page/Page column 94
[9] MedChemComm, 2018, vol. 9, # 2, p. 275 - 281
[10] Journal of Organic Chemistry, 2011, vol. 76, # 3, p. 749 - 759
[11] Patent: WO2006/2981, 2006, A1, . Location in patent: Page/Page column 96-97
[12] Journal of Medicinal Chemistry, 2012, vol. 55, # 22, p. 9467 - 9491
[13] Patent: US2017/313683, 2017, A1, . Location in patent: Paragraph 0289-0290
[14] Organometallics, 2017, vol. 36, # 4, p. 767 - 776
[15] Chemistry - A European Journal, 2016, vol. 22, # 13, p. 4400 - 4404
[16] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 11, p. 5952 - 5961
[17] Journal of Medicinal Chemistry, 2013, vol. 56, # 7, p. 2813 - 2827
[18] Journal of the American Chemical Society, 2011, vol. 133, # 10, p. 3312 - 3315
[19] Patent: EP2548864, 2013, A1, . Location in patent: Paragraph 0319; 0320
[20] Patent: WO2013/14102, 2013, A1, . Location in patent: Page/Page column 64; 65
[21] Helvetica Chimica Acta, 2006, vol. 89, # 5, p. 936 - 946
[22] Organic Letters, 2013, vol. 15, # 1, p. 112 - 115
[23] Organic Letters, 2013, vol. 15, # 24, p. 6190 - 6193
[24] European Journal of Medicinal Chemistry, 1990, vol. 25, # 2, p. 191 - 196
[25] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 8, p. 2342 - 2346
[26] Journal of the American Chemical Society, 2007, vol. 129, # 26, p. 8328 - 8332
[27] Patent: US2007/112011, 2007, A1,
[28] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 22, p. 6134 - 6137
[29] Patent: WO2008/77138, 2008, A1, . Location in patent: Page/Page column 65
[30] Journal of Medicinal Chemistry, 2009, vol. 52, # 7, p. 1853 - 1863
[31] Organic Letters, 2010, vol. 12, # 21, p. 4956 - 4959
[32] European Journal of Medicinal Chemistry, 2010, vol. 45, # 2, p. 588 - 597
[33] Bioorganic and Medicinal Chemistry, 2009, vol. 17, # 17, p. 6422 - 6431
[34] Chemical Communications, 2011, vol. 47, # 46, p. 12553 - 12555
[35] Patent: WO2011/103546, 2011, A1, . Location in patent: Page/Page column 66
[36] Organic Letters, 2012, vol. 14, # 16, p. 4130 - 4133
[37] Chemical Communications, 2012, vol. 48, # 89, p. 11023 - 11025
[38] Chemical Communications, 2014, vol. 50, # 81, p. 12181 - 12184
[39] Journal of Medicinal Chemistry, 2013, vol. 56, # 17, p. 7060 - 7072
[40] Current Medicinal Chemistry, 2014, vol. 21, # 14, p. 1654 - 1666
[41] Journal of the American Chemical Society, 2014, vol. 136, # 29, p. 10266 - 10269
[42] Marine Drugs, 2015, vol. 13, # 1, p. 460 - 492
[43] Patent: WO2016/180537, 2016, A1, . Location in patent: Page/Page column 132
[44] Angewandte Chemie - International Edition, 2016, vol. 55, # 40, p. 12219 - 12223[45] Angew. Chem., 2016, vol. 128, p. 12407 - 12411,5
[46] Marine Drugs, 2016, vol. 14, # 12,
[47] Chemical Communications, 2017, vol. 53, # 33, p. 4593 - 4596
[48] Organic and Biomolecular Chemistry, 2017, vol. 15, # 28, p. 5904 - 5907
[49] Chemical Communications, 2017, vol. 53, # 92, p. 12422 - 12425
[50] Organic Letters, 2018, vol. 20, # 16, p. 4898 - 4901
7
[ 10075-50-0 ]
[ 616-38-6 ]
[ 10075-52-2 ]
Yield
Reaction Conditions
Operation in experiment
99%
With 1,4-diaza-bicyclo[2.2.2]octane In DMF (N,N-dimethyl-formamide) at 90 - 95℃; for 5 h;
To a solution of 5-bromoindole (1.0 g, 5.10 mmol) in DMC (10 ML), DABCO (0.057 g, 0.51 mmol) is added followed by DMF (1 ML).The resulting solution is heated to 90-95° C. for 5 h.The reaction is cooled to RT, and diluted with ethyl acetate (EtOAc, 50 ML) and H2O (50 ML).The organic layer is separated and washed in sequence with H2O (50 ML), 10percent aqueous citric acid (2*50 ML) and H2O (4*50 ML).The organic layer is dried over anhydrous sodium sulfate (Na2SO4), filtered and concentrated under vacuum to give 5-bromo-1-methylindole (about 1.06 g, 99percent) as a golden colored solid: 1H NMR (CDCl3) δ 7.72 (d, 1H), 7.26 (dd, 1H), 7.13 (d, 1H), 7.00 (d, 1H), 6.39 (d, 1H), 3.71 (s, 3H); 13C NMR (CDCl3) δ 135.3, 130.1, 129.9, 124.2, 123.2, 112.6, 110.6, 100.5, 32.9; MS m/z 209 [M+1]+.
Reference:
[1] Patent: US2004/59131, 2004, A1, . Location in patent: Page 5
[2] Organic Process Research and Development, 2001, vol. 5, # 6, p. 604 - 608
[3] RSC Advances, 2014, vol. 4, # 91, p. 50271 - 50276
[4] Synthetic Communications, 2012, vol. 42, # 1, p. 128 - 135
[5] Patent: US6326501, 2001, B1,
[6] Asian Journal of Chemistry, 2018, vol. 30, # 6, p. 1201 - 1204
[7] Chemical Papers, 2018, vol. 72, # 6, p. 1369 - 1378
8
[ 10075-50-0 ]
[ 74-83-9 ]
[ 10075-52-2 ]
Yield
Reaction Conditions
Operation in experiment
100%
Stage #1: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0 - 20℃; for 0.5 h; Stage #2: at 0 - 20℃; for 1 h;
General procedure: Sodium hydride (60percent in mineral oil, 1.10 equiv) was suspended in DMF [0.6 M]. 1H-Indole (1.00 equiv)was solubilized in DMF [1.0 M] and added to the suspension at 0 °C. The mixture was stirred at rt for 30min. Bromoalkyl or TMSCl (1.50 equiv) was diluted in DMF [3.0 M] and added to the solution at 0 °C.The mixture was stirred at rt for 1 hour. The solution was quenched with water (20 mL) and extractedthree times with EtOAc (10 mL). The organic layers were combined, dried over MgSO4 and concentratedunder reduced pressure. The liquid was filtered through a 5 cm pad of silica with 100percent pentane or Et2O toafforded the title compound.
81%
Stage #1: With sodium hydride In N,N-dimethyl-formamide at 0℃; for 0.166667 h; Stage #2: at 0 - 20℃; for 2 h;
General procedure: NaH (6.1 mmol, 1.2 equiv) was slowly added to a solution of 5-bromoindole/indazole (5.1 mmol,1 equiv) in DMF (10 mL) at 0 °C. The mixture was stirred at 0 °C for 10 min then the alkylhalide (6.1 mmol, 1.2 equiv) was added at the same temperature. The reaction mixture wascontinued to stir at rt for 2 h. When the reaction was complete [TLC (EtOAc/hexane 1:5)], themixture was extracted with EtOAc (2 × 20 mL) and washed with water (2 × 20 mL). The organiclayer was separated, dried over anhydrous Na2SO4 and the solvent removed under reducedpressure. The residue obtained was purified by flash column chromatography [silica gel (230–400 mesh; Merck), EtOAc/hexane 1:5].
Reference:
[1] Organic Letters, 2018, vol. 20, # 5, p. 1473 - 1476
[2] Beilstein Journal of Organic Chemistry, 2018, vol. 14, p. 1208 - 1214
[3] Beilstein Journal of Organic Chemistry, 2016, vol. 12, p. 2893 - 2897
9
[ 10075-50-0 ]
[ 10075-52-2 ]
Reference:
[1] Patent: US5252732, 1993, A,
[2] Patent: US5310901, 1994, A,
[3] Chemical Communications, 2013, vol. 49, # 23, p. 2368 - 2370
[4] Patent: US5349061, 1994, A,
[5] Patent: US2010/144726, 2010, A1,
10
[ 10075-50-0 ]
[ 80-48-8 ]
[ 10075-52-2 ]
Reference:
[1] Patent: US6017945, 2000, A,
11
[ 10075-50-0 ]
[ 616-38-6 ]
[ 10075-52-2 ]
Reference:
[1] Journal of Organic Chemistry, 2003, vol. 68, # 5, p. 1954 - 1957
12
[ 10075-50-0 ]
[ 22190-33-6 ]
Yield
Reaction Conditions
Operation in experiment
71%
at 10 - 20℃; for 3.33333 h;
Example 28Preparation of tert-butyl 5-vinylindoline-1-carboxylate (BI10) Step 1. 5-Bromo-indoline (BI11); To 5-Bromo-1H-indole (2.5 g, 12.82 mmol) in acetic acid (10.0 mL), NaCNBH3 (2.38 g, 38.46 mmol) was added portion wise at 10° C. over the period of 20 min After that the reaction mixture was stirred at RT for 3 h. The reaction mixture was diluted with water and extracted with diethyl ether. The organic layer was washed with saturated NaHCO3, water and brine solution. The combined ether layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford title compound as a pale yellow semi-solid (1.8 g, 71percent).
71%
at 10 - 20℃; for 3.33333 h;
To 5-Bromo-1H-indole (2.5 g, 12.82 mmol) in acetic acid (10.0 mL), NaCNBH3 (2.38 g, 38.46 mmol) was added portion wise at 10° C. over the period of 20 min. After that the reaction mixture was stirred at ambient temperature for 3 h. The reaction mixture was diluted with water and extracted with diethyl ether. The organic layer was washed with saturated NaHCO3, water and brine solution. The combined ether layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford title compound as a pale yellow semi-solid (1.8 g, 71percent).
71%
at 10 - 20℃; for 3.33333 h;
To 5-Bromo-1H-indole (2.5 g, 12.82 mmol) in acetic acid (10.0 mL), NaCNBIL (2.38 g, 38.46 mmol) was added portion wise at 10 °C over the period of 20 min. After that the reaction mixture was stirred at RT for 3 h. The reaction mixture was diluted with water and extracted with diethyl ether. The organic layer was washed with saturated NaHCC>3, water and brine solution. The combined ether layer was dried over anhydrous Na2S04 and concentrated under reduced pressure to afford title compound as a pale yellow semi-solid (1.8 g, 71percent).
71%
at 10 - 20℃; for 3 h;
To 5-Bromo-1H-indole (2.5 g, 12.82 mmol) in acetic acid (10.0 mL), NaCNBH3 (2.38 g, 38.46 mmol) was added portion wise at 10° C. over the period of 20 min. After that the reaction mixture was stirred at RT for 3 h. The reaction mixture was diluted with water and extracted with diethyl ether. The organic layer was washed with saturated NaHCO3, water and brine solution. The combined ether layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford title compound as a pale yellow semi-solid (1.8 g, 71percent).
71%
at 10 - 20℃; for 3.33333 h;
Example 28 Preparation of tert-Butyl 5-vinylindoline-1-carboxylate (BI10) Step 1. 5-Bromo-indoline (BI11) To 5-Bromo-1H-indole (2.5 g, 12.82 mmol) in acetic acid (10.0 mL), NaCNBH3 (2.38 g, 38.46 mmol) was added portion wise at 10° C. over the period of 20 min. After that the reaction mixture was stirred at ambient temperature for 3 h. The reaction mixture was diluted with water and extracted with diethyl ether. The organic layer was washed with saturated NaHCO3, water and brine solution. The combined ether layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford title compound as a pale yellow semi-solid (1.8 g, 71percent).
38%
at 0 - 20℃; for 16 h;
10 mL of acetic acid was added to 5-bromoindole (2.5 g, 12.8 mmol) and cooled to 0°C. Sodium triacetoxyborohydride(2.38 g, 37.9 mmol) was added thereto, and the mixture was stirred at room temperature for 16 hours. Afteraddition of water, the reaction solution was extracted with Et2O, and the organic layer washed with sodium bicarbonateaqueous solution. The organic layer was dried with MgSO4 and purified by column chromatography to obtain the titlecompound (0.95 g, 38 percent).1H-NMR (CDCl3) δ 7.19 (1H, d), 7.09 (1H, dd), 6.50 (1H, d), 3.74 (1H, brs), 3.56 (2H, t), 3.02 (2H, t)
Reference:
[1] Organic Letters, 2013, vol. 15, # 11, p. 2798 - 2801
[2] Chemical Papers, 2016, vol. 70, # 5, p. 635 - 648
[3] Tetrahedron Letters, 2005, vol. 46, # 12, p. 2071 - 2074
[4] Nucleosides, Nucleotides and Nucleic Acids, 2005, vol. 24, # 8, p. 1147 - 1165
[5] Chemical Communications, 2017, vol. 53, # 66, p. 9262 - 9264
[6] Journal of Medicinal Chemistry, 2014, vol. 57, # 6, p. 2462 - 2471
[7] Patent: US2012/329649, 2012, A1, . Location in patent: Page/Page column 46
[8] Patent: WO2014/100163, 2014, A1, . Location in patent: Page/Page column 85
[9] Patent: US2014/171308, 2014, A1, . Location in patent: Paragraph 0412
[10] Patent: WO2014/100206, 2014, A1, . Location in patent: Page/Page column 86
[11] Patent: US2014/171312, 2014, A1, . Location in patent: Paragraph 0577; 0578
[12] Patent: US9211280, 2015, B2, . Location in patent: Page/Page column 85-86
[13] Angewandte Chemie - International Edition, 2016, vol. 55, # 40, p. 12219 - 12223[14] Angew. Chem., 2016, vol. 128, p. 12407 - 12411,5
[15] Patent: EP3239143, 2017, A2, . Location in patent: Paragraph 0288
[16] Patent: WO2005/123748, 2005, A1, . Location in patent: Page/Page column 70
[17] Patent: WO2013/166015, 2013, A1, . Location in patent: Paragraph 0202; 0203
[18] Patent: WO2013/166013, 2013, A1, . Location in patent: Paragraph 0122; 0123; 0124
[19] Organic Letters, 2013, vol. 15, # 22, p. 5662 - 5665
[20] Patent: US2014/171314, 2014, A1, . Location in patent: Page/Page column
[21] RSC Advances, 2015, vol. 5, # 30, p. 23727 - 23736
[22] Chemistry - A European Journal, 2015, vol. 21, # 14, p. 5370 - 5379
[23] Journal of Organic Chemistry, 2016, vol. 81, # 2, p. 396 - 403
[24] Chemical Communications, 2017, vol. 53, # 82, p. 11368 - 11371
[25] Journal of Organic Chemistry, 2018, vol. 83, # 4, p. 2425 - 2437
[26] Organic and Biomolecular Chemistry, 2018, vol. 16, # 32, p. 5889 - 5898
[27] Chemical Communications, 2018, vol. 54, # 20, p. 2494 - 2497
13
[ 10075-50-0 ]
[ 124-38-9 ]
[ 1670-81-1 ]
Yield
Reaction Conditions
Operation in experiment
65%
Stage #1: With isopropylmagnesium chloride In tetrahydrofuran at 0℃; for 0.166667 h; Stage #2: With n-butyllithium In tetrahydrofuran; hexane at -20℃; for 0.5 h; Stage #3: at -20℃; for 0.5 h;
To a solution of 5-Bromo-1H-indole (0.86 g, 4.4 mmol, 1.0 equiv.) in dryTHF (20 mL) at 0 C was added a 2 M solution of i-PrMgCl in THF (2.2 mL, 4.4 mmol, 1 equiv.) during5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5Msolution ofn-BuLi in hexanes (3.5 mL, 8.8 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintainingthe temperature below 20 C. The resulting mixture was stirred at that temperature for 0.5 h, dry CO2(0.2 g, 4.4 mmol, 1.0 equiv.) was added to 20 C. The resulting mixture was warmed to 20 C in0.5 h and quenched with water (6 mL). After stirring the mixture below for 10 min, the phases wereseparated and the water phase was extracted one additional time with ethyl acetate. The resultingsuspension was allowed to reach room temperature and fitered through a 0.5 1 cm pad of silicagel eluted with 10 mL of ethyl acetate. The ®ltrate was concentrated and the residue was puri®ed byflash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 3:1) to afford product 3das off-white solid, 0.46 g (yield: 65percent), m.p.: 210–214 C. 1H-NMR (600 MHz, DMSO) 12.39 (s, 1H),11.46 (s, 1H), 8.25 (s, 1H), 7.72 (dd, J = 8.5, 1.5 Hz, 1H), 7.45 (dd, J = 8.4, 5.7 Hz, 2H), 6.57 (s, 1H).13C-NMR (151 MHz, DMSO) 168.90, 138.80, 127.64, 127.35, 123.28, 122.67, 121.87, 111.57, 102.93.
Reference:
[1] Molecules, 2017, vol. 22, # 11,
[2] Journal of the American Chemical Society, 2017, vol. 139, # 28, p. 9467 - 9470
Reference:
[1] Chinese Chemical Letters, 2010, vol. 21, # 12, p. 1407 - 1410
[2] Journal of Medicinal Chemistry, 2014, vol. 57, # 6, p. 2692 - 2703
16
[ 10075-50-0 ]
[ 4692-98-2 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2013, vol. 11, # 43, p. 7455 - 7457
17
[ 10075-50-0 ]
[ 1189-71-5 ]
[ 90271-86-6 ]
Yield
Reaction Conditions
Operation in experiment
91%
Stage #1: at 0℃; for 2 h; Stage #2: With N,N-dimethyl-formamide In acetonitrile at 0℃; for 2 h;
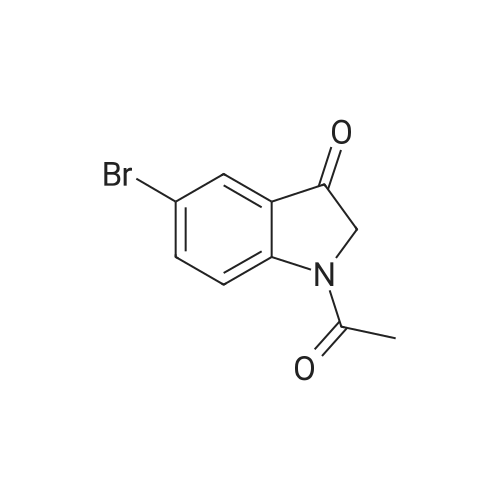
General procedure: To a solution of the appropriate indoles 3a–d (6.8 mmol) in anhydrous acetonitrile (6.0 mL),chlorosulfonyl isocyanate (CSI) (0.63 mL, 7.25 mmol) was added dropwise at 0 °C and the reactionmixture was stirred at 0 °C for 2 h. Anhydrous dimethylformamide (DMF) (1.3 mL, 170.0 mmol)was added dropwise and the mixture was stirred at 0 C for 2 h. The mixture was poured intoice-water and the obtained precipitate was filtered off, dried (anhydrous Na2SO4) and purified bycolumn chromatography using petroleum ether/ethyl acetate (40/60) (for 4b–d) or ethyl acetate (for4a) as eluent.
Reference:
[1] Marine Drugs, 2018, vol. 16, # 8,
18
[ 10075-50-0 ]
[ 75-52-5 ]
[ 90271-86-6 ]
Yield
Reaction Conditions
Operation in experiment
42%
at 90℃; for 36 h;
General procedure: In the same manner as in Example 1 except that the reaction was carried out under the conditions shown in Tables 1 and 2, The compound (3105) was obtained in an isolated yield of 42percent.
Reference:
[1] Advanced Synthesis and Catalysis, 2014, vol. 356, # 2-3, p. 347 - 352
[2] Patent: JP5754768, 2015, B2, . Location in patent: Paragraph 0077; 0078
19
[ 10075-50-0 ]
[ 90271-86-6 ]
Reference:
[1] Chinese Chemical Letters, 2012, vol. 23, # 8, p. 903 - 906
20
[ 10075-50-0 ]
[ 110-18-9 ]
[ 90271-86-6 ]
Reference:
[1] Chemical Communications, 2014, vol. 50, # 18, p. 2315 - 2317
Reference:
[1] Bioorganic and Medicinal Chemistry, 2000, vol. 8, # 2, p. 363 - 371
23
[ 10075-50-0 ]
[ 40432-84-6 ]
Reference:
[1] Tetrahedron Letters, 1994, vol. 35, # 19, p. 3013 - 3016
[2] Organic and Biomolecular Chemistry, 2012, vol. 10, # 10, p. 2101 - 2112
[3] Journal of the American Chemical Society, 2012, vol. 134, # 26, p. 10815 - 10818
24
[ 10075-50-0 ]
[ 1609-47-8 ]
[ 32996-16-0 ]
Reference:
[1] Synthesis, 2004, # 4, p. 568 - 572
25
[ 10075-50-0 ]
[ 124-38-9 ]
[ 10406-06-1 ]
Yield
Reaction Conditions
Operation in experiment
96%
With lithium tert-butoxide In N,N-dimethyl-formamide at 100℃; for 24 h;
General procedure: In a dried two-necked test tube was charged with LiOtBu (160 mg, 2.00 mmol) and indole 1a (23.4 mg, 0.4 mmol). The reaction vessel was evacuated under high vacuum and the atmosphere was replace with a balloon of CO2. Then DMF (2 mL) was added and the mixture was stirred for 24 h at 100°C. Then the result mixture was cooled and carefully quenched with a solution of HCl (2 N) and extracted with EtOAc (5x). The combined organic layers were washed with water (2x), brine (1x) and dry over MgSO4. The dried organics were concentrated under reduce pressure and the residue was purified by preparative TLC (hexane:acetone = 1:1) to afford the desired product 2a (153.0 mg, 95percent) as a white solid.
Reference:
[1] Organic and Biomolecular Chemistry, 2012, vol. 10, # 10, p. 2101 - 2112
37
[ 10075-50-0 ]
[ 5419-55-6 ]
[ 144104-59-6 ]
Yield
Reaction Conditions
Operation in experiment
25%
Stage #1: With sodium hydride In tetrahydrofuran at 0℃; for 0.25 h; Inert atmosphere Stage #2: With n-butyllithium In tetrahydrofuran; hexane at 0℃; for 0.166667 h; Inert atmosphere Stage #3: at 0 - 20℃; Inert atmosphere
A dry and nitrogen-flushed three-necked flask, equipped with a magnetic stirrer and a septum, was charged with NaH (1.02 g, 42.5 mmol) in dry THF (80 mL). The 5-bromoindole (5.0 g, 25.5 mmol) was added dropwise to the solution under an N2 atmosphere at 0 °C, and the reaction mixture was stirred for 15 min. After that the n-BuLi (2.5 M in hexane, 15 mL, 37.5 mmol) was slowly added to the reaction mixture, and then the mixture was stirred for 10 mins. Next the triisopropyl borate (22 mL, 51 mmol) was added dropwise to the mixture. When the addition was complete, the mixture was warmed to room temperature overnight and stirred for 2 hours. The reaction mixture was quenched with saturated NH4Cl and stirred for 30 min. The organic layer was separated and the aqueous layer was extracted with EtOAc. The combined organic layer was washed with water and brine, and dried over Na2SO4. After completely removing the solvent, the residue was treated with EtOAc/petroleum ether and filtered to afford 1H-indol-5-ylboronic acid (b1) as colorless solid (1.02 g, 25percent yield). 21H-NMR (DMSO-d6) δ 11.1 (s, 1H), 8.30 (s, 1H), 7.81 (d, 1H, J=8.1 Hz), 7.47 (d, 1H, J=8.1 Hz), 7.34 (s, 1H).13C-NMR (100 MHz, (DMSO-d6) δ 138.0, 128.0, 127.5, 127.4, 127.3, 125.4, 111.0, 102.0.
Reference:
[1] Patent: WO2006/51851, 2006, A1, . Location in patent: Page/Page column 71-72
[2] Journal of Organic Chemistry, 2004, vol. 69, # 20, p. 6812 - 6820
[3] Journal of Organic Chemistry, 2007, vol. 72, # 3, p. 1047 - 1050
[4] Organic Letters, 2012, vol. 14, # 18, p. 4814 - 4817,4
[5] Journal of Organic Chemistry, 2013, vol. 78, # 13, p. 6427 - 6439
39
[ 10075-50-0 ]
[ 688-74-4 ]
[ 144104-59-6 ]
Reference:
[1] Patent: US6307056, 2001, B1,
40
[ 10075-50-0 ]
[ 5419-55-6 ]
[ 144104-59-6 ]
Reference:
[1] Patent: EP1820799, 2007, A1,
41
[ 10075-50-0 ]
[ 13675-18-8 ]
[ 144104-59-6 ]
Reference:
[1] Organic Process Research and Development, 2017, vol. 21, # 11, p. 1859 - 1863
42
[ 10075-50-0 ]
[ 75-03-6 ]
[ 195253-49-7 ]
Yield
Reaction Conditions
Operation in experiment
89%
Stage #1: With sodium hydride In DMF (N,N-dimethyl-formamide) at 80℃; for 0.5 h; Stage #2: at 80℃; for 2 h;
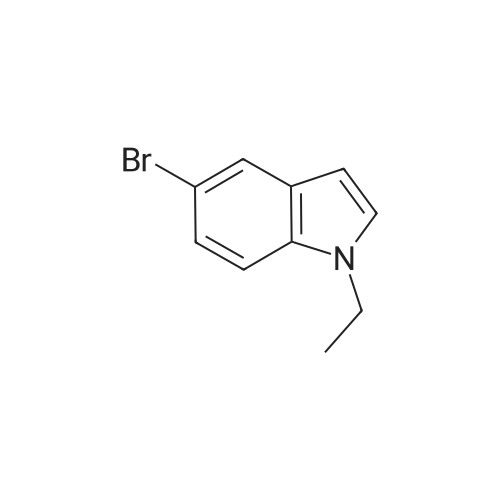
Example 15 5-Bromo-3-(2,4-dichlorobenzoyl)-1-ethyl-1H-indole [0380] 1a) 5-Bromo-1-ethyl-1H-indole [0381] In 75 mL of anhydrous dimethylformamide, place 3.06 g (76.5 mmol) of sodium hydride in 60percent suspension in mineral oil. Stir and heat the mixture to 80° C. Add progressively, using a spatula, 5.0 g (25.5 mmol) of 5-bromo-1H-indole. Continue the heating for 30 minutes (until no further hydrogen is evolved). Cool the mixture to ambient temperature and add 4 mL (51 mmol) of iodoethane. Heat the mixture again to 80° C. for 2 hours. Hydrolyse the reaction medium. Extract with dichloromethane, dry the organic phase over anhydrous sodium sulfate and evaporate. Purify the evaporation residue by chromatography on silica gel with elution by dichloromethane (Int. 49). [0382] Yield: 89percent [0383] Yellow oil
Reference:
[1] Archiv der Pharmazie, 1997, vol. 330, # 5, p. 141 - 145
[2] Patent: US2004/67998, 2004, A1, . Location in patent: Page/Page column 18
43
[ 10075-50-0 ]
[ 74-96-4 ]
[ 195253-49-7 ]
Reference:
[1] Tetrahedron Letters, 2017, vol. 58, # 1, p. 35 - 42
[2] MedChemComm, 2018, vol. 9, # 2, p. 275 - 281
[3] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 11, p. 5952 - 5961
[4] European Journal of Medicinal Chemistry, 2010, vol. 45, # 2, p. 588 - 597
[5] Bioorganic and Medicinal Chemistry, 2009, vol. 17, # 17, p. 6422 - 6431
44
[ 75-21-8 ]
[ 10075-50-0 ]
[ 148366-28-3 ]
Reference:
[1] Patent: US5310901, 1994, A,
[2] Patent: US5352783, 1994, A,
[3] Patent: US5252732, 1993, A,
[4] Patent: US5349061, 1994, A,
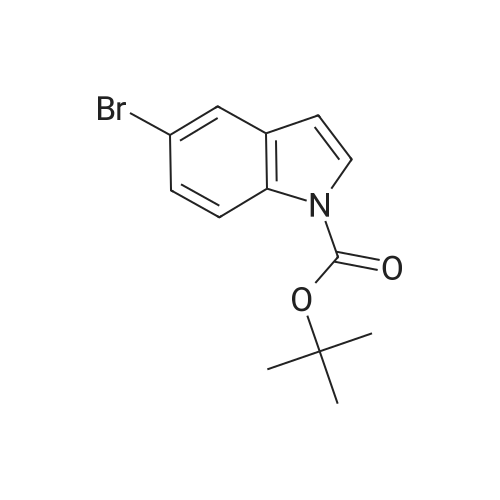
A solution of 5-bromo-indole (5.0 g, 25.5 mmol), di-tert-butyl dicarbonate (7.6 mL, 33.1 mmol) and 4-dimethylaminopyridine (0.15 g, 1.23 mmol) in acetonitrile (50 mL) was stirred at room temperature for 3 hr. An aqueous citric acid solution was added to the reaction solution, and the mixture was extracted with ethyl acetate. The extract was washed with aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to give the title compound (7.8 g, yield 100percent) as a pale-yellow oil.
99%
With dmap In acetonitrile at 23℃; for 0.5 h;
Example 12. Synthesis of N-Boc-5-bromoindole (SlO)99percent S10To 5-bromoindole (196 mg, 1.00 mmol, 1.00 equiv) in acetonitrile (2.0 niL) at 23 °C was added di-tert-buty\\ dicarbonate (276 rnL, 1.20 mmol, 1.20 equiv) and A- dimethylaminopyridine (12.0 mg, 0.100 mmol, 10.0 molpercent). After stirring for 30 min at 23 0C, the reaction mixture was concentrated in vacuo. The residue was purified by chromatography on silica gel eluting with hexanes/EtOAc 30:1 (v/v) to afford 293 mg of the title compound as a colorless solid (99percent yield).R/= 0.35 (hexanes/EtOAc 30:1 (v/v)). NMR Spectroscopy: 1H NMR (400 MHz, CDCl3, 23 0C, δ): 8.02 (d, / = 8.8 Hz, IH), 7.69 (d, / = 2.0 Hz, IH), 7.58 (d, / = 3.6 Hz, IH), 7.39 (dd, /= 8.8 Hz, 2.0 Hz, IH), 6.50 (d, / = 3.6 Hz, IH), 1.67 (s, 9H). 13C NMR (100 MHz, CDCl3, 23 0C, δ): 149.40, 133.90, 132.22, 127.00, 123.51, 116.54, 115.94, 106.45, 84.12, 28.14. (Note: Only ten peaks were observed probably due to accidental overlap of two peaks)
97%
With dmap; triethylamine In dichloromethane at 20℃; for 16 h;
[0520] To a solution of compound 51 (30 g, 0.15 mol) in DCM (300 mL) was added Et3N (64 mL, 0.46mo1), DMAP (5.6g, 0.046 mol), and then Boc2O (50 g, 0.23 mol) in portions at 0°C. After addition, the reaction mixture was stirred at room temperature for l6hrs. The mixture was diluted with DCM (200 mL) and washed with water and brine, dried over anhydrous Na2504, filtered and concentrated to dryness. The residue was purified by column chromatography on silica gel (eluted with Petroleum ether: Ethyl acetate = 50: 1) to give compound S2 (44 g, 97 percent yield) as a white solid. LC/MS (ESI) m/z: 240 (M-56+H).
97%
With dmap; triethylamine In dichloromethane at 20℃; for 16 h;
A mixture of 5-bromo-lH-indole (10.0 g, 51.0 mmol), Boc20 (16.7 g, 76.5 mmol), TEA (14.2 mL, 102.0 mmol) and DMAP (622 mg, 5.1 mmol) in DCM (150 mL) was stirred at room temperature for 16hrs. The mixture was evaporated under reduced pressure and the residue was purified by chromatography on silica gel (PE: EtOAc=50: l) to give the title compound (14.6 g, 97.0 percent yield) as white solid.
97%
With dmap; triethylamine In dichloromethane at 0 - 20℃; for 16 h; Inert atmosphere
[0572] To a solution of compound 116 (30 g, 0.15 mol) in DCM (300 mL) was added Et3N (64 mL, 0.46mol), DMAP (5.6 g, 0.046 mol), then Boc2O (50 g, 0.23 mol) was added in portions at 0 oC. After addition, the reaction was stirred at room temperature for 16 hrs. The mixture was diluted with DCM (200 mL) and washed with water and brine, dried over anhydrous Na2SO4, filtered and concentrated to dryness. The residue was purified by column chromatography on silica gel (eluted with Petroleum ether: Ethyl acetate = 50: 1) to give the title compound (44 g, 97 percent yield) as white solid. LC/MS (ESI) m/z: 240 (M-56+H)+.
97%
With dmap; triethylamine In dichloromethane at 0 - 20℃; for 16 h;
To a solution of compound S1 (30 g, 0.15 mol) in DCM (300 mL) at 0° C. was added Et3N (64 mL, 0.46 mol), DMAP (5.6 g, 0.046 mol). This was followed by addition of Boc2O (50 g, 0.23 mol) in portions. The reaction mixture was stirred at room temperature for 16 hrs. The mixture was diluted with DCM (200 mL) and washed with water and brine, dried over anhydrous Na2SO4, filtered and concentrated to dryness. The residue was purified by column chromatography on silica gel (eluted with petroleum ether: ethyl acetate=50:1) to give the title compound (44 g, 97percent yield) as white solid. LC/MS (ESI) m/z: 240 (M-56+H)+.
97%
With dmap; triethylamine In dichloromethane at 0 - 20℃; for 16 h;
To a solution of Scheme 6-7 compound 51 (30 g, 0.15 mol) in DCM (300 mL) was added Et3N (64 mL, 0.46mo1), DMAP (5.6 g, 0.046 mol), then Boc2O (50 g, 0.23 mol) was added in portions at 0 °C. After the addition was complete, the reaction was stirred at room temperature for 16 h. The mixture was diluted with DCM (200 mL), washed with water and brine, dried over anhydrous Na2504, filtered and concentrated to dryness. The residue was purified by column chromatography on silica gel (eluted with petroleum ether: ethyl acetate = 50: 1) to afford Scheme 6-7 compound S2 (44 g, 97 percent yield) as a white solid. LC/MS (ESI) m/z: 240 (M-56+H)t
86%
With dmap In tetrahydrofuran at 20℃; for 16 h;
Preparation of ferf-butyl 5-bromo-1H-indole-1-carboxylate Boc-anhydride (12.8 g, 58.65 mmol) was added to a stirred solution of 5-bromo-I H- indote (5.0 g, 25.50 mmol) in THF (100 mL) at room temperature. DMAP (1 ,24 g: 10.20 mmol) was added portionwise. The reaction mass was stirred at room temperature for 16 hours. After complete consumption of the starting material, THF was evaporated under vacuum. The residue was dissolved in EtOAc, washed with water folfowed by brine solution and dried over anhydrous Na2SO The organic layer was concentrated under reduced pressure to afford the crude product. The crude compel was purified on 100-200 mesh silica-gel eluting with 10percent EtOAc in petroleum ether to obtain an white solid (6.5 g, 86percent). 1H NMPv (400 MHz, CDCI3): δ 8.02 (br d, J - 8.4 Hz, 1 H), 7.69 (d, J - 2.0 Hz, 1 H), 7.58 (d, J = 4.0 Hz, 1 H), 7.39 (dd, J = 8.8 Hz, 2.0 Hz, 1 H), 6.50 (d, J = 3.6 Hz, 1 H), 1.67 (s, 9H). LCMS: m/z 297.0 [M+H]+.
23%
With triethylamine In dichloromethane at 20℃; Heating / reflux
(BoC)2O (23.35 g, 107.11 mmol) was added to a solution of 5-bromo-lH-indole (20 g, 102.04 mmol) in dichloromethane (120 mL). Et3N (10.302 g, 102.00 mmol) was then added, and the resulting solution was stirred at room temperature overnight, then for 6 hours at reflux temperature. The residue was purified by column chromatography using petroleum ether solvent to afford 7.21 g (23percent) of tert- butyl 5-bromo-lH-indole-l -carboxylate as a white solid.
27 g
With dmap In dichloromethane at 0 - 20℃; for 16 h;
1 ,1 -Dimethylethyl 5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-indole-1 - carboxylate(a) 1 ,1 -Dimethylethyl 5-bromo-1 H-indole-1 -carboxylate5-Bromoindole (20 g) and DMAP (1 g) were dissolved in DCM (200 ml.) and cooled to 0 °C. To this was added di-t-butyl dicarbonate (29 g) in portions. The reaction was allowed to warm to room temperature and was stirred at RT for 16 hrs. The reaction mixture was diluted with water and the organic layer was separated. The aqueous layer was extracted once with DCM and the combined organic layers were dried and evaporated to dryness. The crude product was purified by flash chromatography on silica to afford 27 g of the titled compound.
Reference:
[1] Patent: EP1731505, 2006, A1, . Location in patent: Page/Page column 29-30
[2] Journal of the American Chemical Society, [3] Journal of the American Chemical Society, 2009, vol. 131, p. 1662 - 1663
[4] Patent: WO2010/59943, 2010, A2, . Location in patent: Page/Page column 36
[5] Synthesis, 2009, # 21, p. 3617 - 3632
[6] Patent: WO2017/35360, 2017, A1, . Location in patent: Paragraph 0520
[7] Patent: WO2017/35349, 2017, A1, . Location in patent: Paragraph 0638
[8] Patent: WO2017/35357, 2017, A1, . Location in patent: Paragraph 0461; 0572
[9] Patent: WO2017/35353, 2017, A1, . Location in patent: Paragraph 0710
[10] Patent: WO2017/35417, 2017, A1, . Location in patent: Paragraph 0630; 0652
[11] Angewandte Chemie, International Edition, 2009, vol. 48, # 23, p. 4235 - 4238
[12] Tetrahedron, 2009, vol. 65, # 34, p. 6877 - 6881
[13] Bioorganic and Medicinal Chemistry, 2011, vol. 19, # 7, p. 2156 - 2167
[14] Tetrahedron, 2000, vol. 56, # 43, p. 8473 - 8480
[15] Journal of the American Chemical Society, 2017, vol. 139, # 14, p. 5003 - 5006
[16] Journal of Organic Chemistry, 2004, vol. 69, # 20, p. 6812 - 6820
[17] Patent: WO2015/74123, 2015, A1, . Location in patent: Page/Page column 64
[18] Synthesis, 2008, # 5, p. 707 - 710
[19] Chemical Communications, 2012, vol. 48, # 26, p. 3239 - 3241
[20] Heterocycles, 2018, vol. 96, # 6, p. 1067 - 1074
[21] Synlett, 2008, # 2, p. 294 - 296
[22] Patent: WO2007/98418, 2007, A1, . Location in patent: Page/Page column 98
[23] Journal of Organic Chemistry, 2002, vol. 67, # 21, p. 7551 - 7552
[24] Patent: WO2013/28445, 2013, A1, . Location in patent: Page/Page column 56
[25] Journal of Medicinal Chemistry, 2013, vol. 56, # 17, p. 7060 - 7072
[26] Current Medicinal Chemistry, 2014, vol. 21, # 14, p. 1654 - 1666
[27] Marine Drugs, 2015, vol. 13, # 1, p. 460 - 492
[28] ACS Medicinal Chemistry Letters, 2018, vol. 9, # 2, p. 131 - 136
50
[ 10075-50-0 ]
[ 51300-90-4 ]
[ 182344-70-3 ]
Reference:
[1] Marine Drugs, 2016, vol. 14, # 12,
51
[ 10075-50-0 ]
[ 400071-95-6 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2001, vol. 36, # 6, p. 545 - 553
[2] Patent: EP2548864, 2013, A1,
[3] Patent: WO2013/14102, 2013, A1,
[4] Tetrahedron, 2016, vol. 72, # 5, p. 734 - 745
52
[ 10075-50-0 ]
[ 325800-39-3 ]
Reference:
[1] Chemical Papers, 2016, vol. 70, # 5, p. 635 - 648
Reference:
[1] Journal of Organic Chemistry, 2002, vol. 67, # 21, p. 7551 - 7552
55
[ 10075-50-0 ]
[ 61350-62-7 ]
[ 143322-56-9 ]
Yield
Reaction Conditions
Operation in experiment
83%
Stage #1: With aluminum (III) chloride In dichloromethane at 0 - 5℃; for 1 h; Stage #2: at 0 - 5℃; for 6 h;
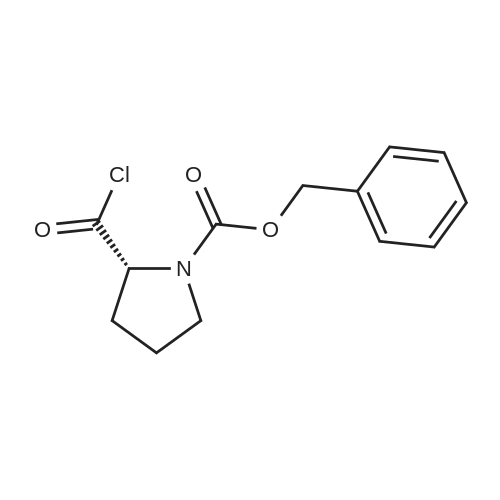
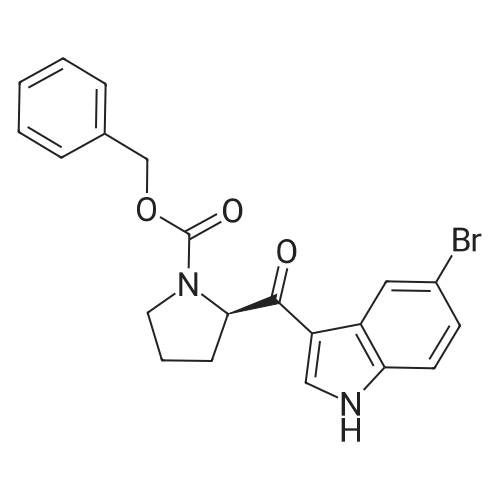
5-bromoindole (15.7 g, 80.4 mmol), dichloromethane (140 mL) was added to a 500 ml four-necked flask,The ice bath was cooled to 0 ° C and the aluminum trichloride solid (16.1 g, 120.6 mmol) was slowly added in portions,The internal temperature control in the 0 ~ 5 , stirring reaction 1h, 0 ~ 5 conditions,A solution of N-benzyloxycarbonyl prolyloyl chloride in Example 1 in dichloromethane was added dropwise,The reaction solution was stirred at 0-5 ° C for 6 h. Ice bath, 90mL saturated ammonium chloride solution slowly drip,First filter, the filtrate layer, the organic phase in turn with saturated sodium bicarbonate, saturated salt water washing,After concentration under reduced pressure, the crude product was purified by recrystallization from a mixed solvent of ethyl acetate and n-heptane (1: 1 by volume), vacuum dried at 60 ° C for 8 hours,(R) -3- (N-benzyloxycarbonylpyrrolidin-2-ylcarbonyl) -5-bromo-lH-indole (14.2 g) as a white solid,The yield was 83percent and the HPLC purity was 99.2percent.
Stage #1: With tert-butylmagnesium chloride In diethyl ether at 0 - 20℃; for 0.75 h;
N-Benzyloxycarbonyl-R-proline acid chloride from the above reaction was dissolved in anhydrous diethyl ether (30 mL) and added at 0 °C to a solution of 5-bromoindole (2.9 g, 15.0 mmol) and t-butylmagnesium chloride (2M solution in diethyl ether, 8.3 mL, 16.5 mmol) in anhydrous diethyl ether (30 mL). The resulting mixture was stirred at room temperature under argon for 45 minutes and then ethyl acetate (150 mL) and saturated sodium bicarbonate (30 mL) were added. The organic layer was dried and evaporated under reduced pressure to provide a yellow oil. The title compound was crystallized using hexane/ethyl acetate (9:1) to provide a white solid (3.07 g, 72percent). mp 95-96 °C.
Reference:
[1] Patent: US5545644, 1996, A,
[2] Patent: US5559129, 1996, A,
[3] Patent: US5559246, 1996, A,
[4] Patent: US5578612, 1996, A,
[5] Patent: US5607951, 1997, A,
59
[ 10075-50-0 ]
[ 925-90-6 ]
[ 144-55-8 ]
[ 61350-62-7 ]
[ 143322-56-9 ]
Reference:
[1] Patent: US5545644, 1996, A,
[2] Patent: US5559129, 1996, A,
[3] Patent: US5559246, 1996, A,
[4] Patent: US5578612, 1996, A,
[5] Patent: US5607951, 1997, A,
[6] Patent: US5639779, 1997, A,
60
[ 10075-50-0 ]
[ 73183-34-3 ]
[ 269410-24-4 ]
Yield
Reaction Conditions
Operation in experiment
90%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane for 8 h; Inert atmosphere; Reflux
Into a 500 ml single-mouth flask, successively add 5-bromoindole (20.00g, 0 . 1mol), bis(pinacolato)diboron (51.80g, 0 . 2mmol), potassium acetate (22.80g, 0 . 2mol), [ 1,1 '-bis (diphenylphosphino) ferrocene] palladium dichloride (1.50g), and 350 ml dioxane. Under the protection of nitrogen, reflux for 8h. Cooling latter turns on lathe steams removing dioxane, with 200 ml distilled water dichloromethane is used for extraction after washing 3 times. Re-crystallization with methanol-methylene chloride to obtain the product 21.8g, the yield is 90percent.
83%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 100℃; for 5.33333 h; Inert atmosphere
5-Bromoindole (2 g, 10.2 nmol) and Bis(pinacolato)diboron (4 g, 19.2 mmol) was dissolved in 1,4-dioxane and then PdCl2(dppf) (524 mg, 0.72 mmol) and potassium acetate(3 g, 30.6 mmol) were added. The suspension was bubbled with nitrogen for 20 min and then stirred at 100 °C for 5 h. After the mixture was cooled at room temperature and quenched with water. Then the solution was extracted with ethyl acetate (20 mL × 3), and the combined organic layer was washed by saturated sodium chloride solution for three times, dried over anhydrous Na2SO4 and concentrated under reduced pressure. Finally, the residue was puried by silica gel chromatography to get white solid a. HPLC purity: 96.7percent. Yield: 83percent. 13C NMR (100MHz, DMSO-d6): δ 138.34(1C, C-8), 128.32(1C, C-3), 127.77(1C, C-5), 127.29(1C, C-1), 126.94(1C, C-4), 123.79(1C, C-6), 111.27(1C, C-7), 102.03(1C, C-2), 83.42(2C, - C-O), 25.06(4C, -CH3). HR-MS (ESI) calcd for C14H18BNO2 [M+ Na] +: 266.1323; found: 266.1393.
75%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 130℃; for 12 h;
Under a nitrogen flow 5-bromo-1H-indole (100.0 g, 510.1 mmol), 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi (1,3 ,2-dioxaborolane) (142.5 g, 561.1 mmol), Pd(dppf)Cl2 (44.7 g, 51.0 mmol), KOAc (144.1 g, 1.52mol) and 1,4-dioxane (2500 ml) and the mixture was stirred at 130 °C for 12 hours. After the reaction was terminated by the removal of water and then extracted with ethyl acetate MgSO4, and purified by column chromatography. (Hexane: EA = 8: 1 (v / v)) to give 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (93.0 g, with a yield of 75percent).
72%
With [1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II); potassium acetate In 1,4-dioxane at 130℃; for 12 h; Inert atmosphere
Under a nitrogen flow 5-bromo-1H-indole (25 g, 0.128 mol), 4,4,4 ', 4', 5,5, 5 ', 5'-octamethyl-2,2'-bi (1,3 ,2-dioxaborolane) (48.58 g, 0.191 mol), Pd (dppf) Cl2 (5.2 g, 5 mol), KOAc (37.55g, 0.383mol) and 1,4-dioxane (500 ml) the mixture, and then the 130 in it was stirred for 12 hours.After the reaction was completed, the removal of water in our MgSO4 and then extracted with ethyl acetate, column chromatography (Hexane: ethyl acetate (EA) = 10: 1 (v / v)) to give 5- (4,4,5 a , 5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1H-indole (22.32 g, a yield of 72percent).
72%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 130℃; for 12 h; Inert atmosphere
Under a nitrogen stream, 5-bromo-1H-indole (25g, 0.128mol), 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane)(48.58g, 0.191mol), Pd (dppf) Cl2 (5.2g, 5mol), KOAc (37.55g, 0.383mol), and 1,4-dioxane (500ml) mixed, and the mixture was stirred for 12 hours at 130°C .After completion of the reaction, and extracted with ethyl acetate, water was removed by MgSO4, column chromatography (hexane: EA = 10: 1 (v / v)) purified to at 5- (4,4,5,5-tetra to give methyl 1,3,2-dioxaborolan-2-yl)-lH-indole (22.32g, 72percent yield).
72%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 130℃; for 12 h; Inert atmosphere
Under a nitrogen flow 5-bromo-1H-indole (25 g, 0.128 mol), 4,4,4 ', 4', 5,5, 5 ', 5'-octamethyl-2,2'-bi (1,3 ,2-dioxaborolane) (48.58 g, 0.191 mol), Pd (dppf) Cl2 (5.2 g,5 mol), KOAc (37.55 g, 0.383 mol) and 1,4-dioxane (500 ml) and the mixture 130 ° Cin was stirred for 12 hours. After the reaction was terminated by the removal of water and then extracted with ethylacetate and MgSO4, purified by column chromatography (Hexane: EA = 10: 1 (v / v))purified 5- (4,4,5,5-tetramethyl- by a 1,3,2-dioxaborolan-2-yl) -1H-indole (22.32g, a yield of 72percent).
72%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 130℃; for 12 h; Inert atmosphere
Under a nitrogen flow 5-bromo-1H-indole (25 g, 0.128 mol), 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1,3,2-dioxaborolane) (48.58 g, 0.191 mol), Pd(dppf)Cl2 (5.2 g, 5 mol), KOAc (37.55 g, 0.383 mol) and 1,4-dioxane (500 ml) and the mixture at 130°C , It was stirred for 12 hours. After the reaction was terminated by the removal of water and then extracted with ethyl acetate and MgSO4, purified by column chromatography (Hexane: EA = 10: 1 (v / v)) 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (22.32 g, a yield of 72percent).
72%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 130℃; for 12 h; Inert atmosphere
Under a nitrogen stream, 5-bromo -1H- indole (25g, 0.128mol), 4,4,4 ', 4', 5,5,5 ', 5'- octamethyl-2,2'-bi (1, 3,2-dioxaborolane) (48.58g, 0.191mol), Pd (dppf) Cl2 (5.2g, 5molpercent), KOAc (37.55g, 0.383mol), and 1,4-dioxane (500ml) It was mixed, and the mixture was stirred for 12 hours at 130°C .After completion of the reaction, and extracted with ethyl acetate, water was removed by MgSO4, column chromatography (hexane:: EA = 10 1 (v / v)), 5- (4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole was obtained.(22.32g, 72percent yield).
72%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 130℃; for 12 h; Inert atmosphere
Under the nitrogen air 5-bromo-1H-indole (25 g, 0.128 mol), 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1,3,2-dioxaborolane) (48.58 g, 0.191 mol), pd(dppf)Cl2 (5.2 g, 5 mol), KOAc (37.55 g, 0.383 mol) and 1,4-dioxane (500 ml) were mixed, then it was stirred at 130 °C for 12 hours. After reaction being terminated, then ethyl acetate extraction of MgSO 4 for removing moisture to, column chromatography ( reaction is concluded Hexane: it was refined to the EA = 10:1 (v/v)) for purifying the 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (22.32 g, yield 72percent) were obtained.
72%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 130℃; for 12 h; Inert atmosphere
Under a nitrogen flow 5-bromo-1H-indole ( 25 g, 0.128 mol), 4,4,4 ', 4', 5,5,5 ', 5'-octamethyl-2,2'-bi (1,3,2-dioxaborolane) (48.58 g, 0.191 mol), Pd (dppf) Cl 2 (5.2 g, 5 mol), KOAc (37.55 g, 0.383 mol)and 1,4-dioxane (500 ml) and the mixture 130 °C in was stirred for 12 hours. The reaction was terminated, and then extracted with ethyl acetate MgSO4to remove water to and purified by column chromatography to give 5- (4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl) -1H-indole to obtain a (22.32 g, 72percent yield).
72%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 130℃; for 12 h; Inert atmosphere
synthesis of 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1H- indole Under a nitrogen stream, 5-bromo--1H- indole (25g, 0.128mol) 4,4,4 ', 4', 5,5,5 ', 5'- octamethyl-2,2'-bi (1,3 , 2-dioxaborolane) (mixed 48.58g, 0.191mol), Pd (dppf) Cl2 (5.2g, 5mol), KOAc (37.55g, 0.383mol), and 1,4-dioxane (500ml) after, the mixture was stirred for 12 hours at 130 . After completion of the reaction, and extracted with ethyl acetate, water was removed by MgSO4, column chromatography [hexane: ethyl acetate (EA) = 10: 1 (v / v)] in purified 5- (4,4,5 to give 5-tetramethyl-1,3,2-dioxaborolan-2-yl)-lH-indole (22.32g, 72percent yield).
72%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 130℃; for 12 h; Inert atmosphere
Under nitrogen, 5-bromo-1H-indole (25 g, 0.128 mol), 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1,3,2-dioxaborolane) (48.58 g, 0.191 mol), Pd(dppf)Cl2 (5.2 g, 0.05 mol), and potassium acetate (KOAc) (37.55 g, 0.383 mol) were dissolved in a round-bottomed flask filled with 1,4-dioxane (500 ml). The mixed reactants were stirred for 12 h at 130 °C. After the reaction was terminated, the mixture was extracted with ethyl acetate three times. The organic layer was separated and the moisture was removed by drying over MgSO4. After removing the solvent by evaporation, the crude product was purified by column chromatography with an ethyl acetate and n-hexane (1:10) eluent. Yield 72percent (22.32 g), 1H NMR (400 MHz, DMSO): δ 1.24 (s, 12H), 6.45 (d, 1H), 7.27 (d, 1H), 7.42 (d, 1H), 7.52 (d, 1H), 7.95 (s, 1H), 8.21 (s, 1H). MS (APCI) m/z 244.11 [(M + H)+].
72%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 130℃; for 12 h;
5-bromo-1H-indole 25 g (128 mmol), 4,4,4 ', 4', 5,5, 5 ', 5'-octamethyl-2,2'-bi (1,3,2-dioxaborolane ) 48.58 g (0.191 mmol), Pd (dppf) Cl25.2 g (6.4 mmol), KOAc 37.55 g (383 mmol) and dioxane 500 mL and 130 the mixture heated at reflux 12 hours.Allowed to cool to room temperature and quench the reaction with 500 mL aqueous solution of ammonium chloride to the reaction mixture.Extract the mixture with EA 500 mL, and washed with distilled water.The resulting organic layer was dried over anhydrous MgSO4,and evaporated under reduced pressure to give the desired compound 22.32 g (72percent yield) was purified by a silica gel column chromatography.
72%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 130℃; for 12 h;
(25 g, 0.128 mol), 4,4 ', 4', 5,5,5 ', 5'-octamethyl-2,2'-bi (1,3, Dioxane (500 ml) were mixed and heated at 130 for 12 hours at 130 . The resulting mixture was stirred at 130 for 2 hours to obtain a mixture of d- (2-dioxaborolane) (48.58 g, 0.191 mol), d After completion of the reaction, the reaction mixture was extracted with ethyl acetate, the water was removed with MgSO 4, and the residue was purified by column chromatography to obtain 5- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2 -yl) -1H-indole (22.32 g, yield 72percent).
40%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In N,N-dimethyl-formamide at 130℃; for 12 h; Inert atmosphere
Under a nitrogen stream, 5-bromo-1H-indole (25g, 0.128mol), 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi-(1,3,2-dioxaborolane) (48.58g, 0.191mol), Pd(dppf)Cl2 (5.2g, 5mol), KOAc (37.55g, 0.383mol) and a mixture of DMF (500ml), 12 hours with stirring at 130 °C. After completion of the reaction, and extracted with ethyl acetate, to remove water with MgSO4, is purified by Karamukuro Matogurafi (Hexane: EA = 10: 1 (v / v)) was obtained 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolane-2-yl)-1H-indole (12.43g, 40percent yield).
40%
With palladium bis[bis(diphenylphosphino)ferrocene] dichloride; potassium acetate In N,N-dimethyl-formamide at 130℃; for 12 h; Inert atmosphere
5-bromo-1H-indole (25 g, 0.128 mol) was added under a nitrogen stream, 4,4,4 ', 4', 5,5,5 ', 5'-octamethyl-2,2'-bi (1,3,2-dioxaborolane)(48.58 g, 0.191 mol), Pd (dppf) Cl2 (5.2 g, 5 mol), KOAc (37.55 g, 0.383 mol) andWere mixed DMF (500 ml) And the mixture was stirred at 130 DEG C for 12 hours.After the reaction was completed, the reaction mixture was extracted with ethyl acetate, the water was removed with MgSO4,Purification by column chromatography (Hexane: EA = 10: 1 (v / v)) gave5- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1H-indole(12.43 g, yield 40percent).
280 mg
With tris-(dibenzylideneacetone)dipalladium(0); potassium acetate; XPhos In 1,4-dioxane at 115℃; for 18 h; Inert atmosphere
To a solution of 5-bromo-1H-indole (200 mg, 1.02 mmol) in dioxane (10 mL) were added bis (pinacolato) diboron (309.8 mg, 1.22 mmol) , potassium acetate (300 mg, 3.06 mmol) , X-Phos (48.5 mg, 0.10 mmol) and tris (dibenzylideneacetone) dipalladium (46.7 mg, 0.05 mmol) . The mixture was stirred under N2at 115 for 18 h. After the reaction was completed, the reaction mixture was filtered through a Celite pad. The filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography eluted with PE/EtOAc (v/v) 8/1) to give a yellow solid product (280 mg) .[0781]MS (ESI, pos. ion) m/z: 244.2 [M+1]+.
3.65 g
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In toluene for 16 h; Reflux
Bromide 1-6-a (3.92 g, 20 mmol), boronic acid pinacol ester (6.22 g, 24 mmol),1,1'-bis(diphenylphosphino)-ferrocene-palladium(II)dichloride dichloromethane complex (0.49 g, 0.6 mmol), potassium acetate (5.90 g, 0 mmol) and 79 ml of toluene The reaction was refluxed for 16 hours, cooled, added with 26 ml of water, stirred for 30 minutes, the organic phase was separated, filtered through a short celite bed, and then the organic solvent was evaporated, and the crude product was recrystallized from heptane / toluene; Under argon atmosphere,The obtained solid (3.65 g, 15 mmol), bromobenzene (2.06 g, 14.3 mmol), tetrakis(triphenylphosphine)palladium (0.35 g, 0.3 mmol), toluene (43 ml), aqueous sodium carbonate (2M, 21 ml) The mixture was placed in a flask, and the mixture was refluxed for 8 hours, cooled to room temperature, and extracted with toluene. The organic phase was washed with saturated brine, and then dried, and then purified by column chromatography to afford compound 1-6 3.59 g.
Reference:
[1] Patent: CN105601612, 2016, A, . Location in patent: Paragraph 0051; 0052; 0053
[2] Journal of Organic Chemistry, 2004, vol. 69, # 20, p. 6812 - 6820
[3] Organic Letters, 2012, vol. 14, # 2, p. 600 - 603
[4] Bioorganic and Medicinal Chemistry Letters, 2017, vol. 27, # 17, p. 4150 - 4155
[5] Patent: KR101603387, 2016, B1, . Location in patent: Paragraph 0084-0088; 0143-0146
[6] Patent: KR101571589, 2015, B1, . Location in patent: Paragraph 0172-0176
[7] Patent: JP2015/528445, 2015, A, . Location in patent: Paragraph 0047; 0048
[8] Patent: KR101548040, 2015, B1, . Location in patent: Paragraph 0094-0098
[9] Patent: KR2015/88007, 2015, A, . Location in patent: Paragraph 0095; 0096; 0097
[10] Patent: JP2015/527347, 2015, A, . Location in patent: Paragraph 0052; 0053; 0069; 0074
[11] Patent: KR101577099, 2015, B1, . Location in patent: Paragraph 0168-0172
[12] Patent: KR2015/38811, 2015, A, . Location in patent: Paragraph 0140-0143
[13] Patent: JP2015/530991, 2015, A, . Location in patent: Paragraph 0056-0057
[14] Dyes and Pigments, 2017, vol. 136, p. 529 - 534
[15] Patent: KR101571592, 2015, B1, . Location in patent: Paragraph 0130-0134
[16] Patent: KR101599586, 2016, B1, . Location in patent: Paragraph 0110-0113
[17] Synlett, 2003, # 8, p. 1204 - 1206
[18] Journal of the American Chemical Society, 2017, vol. 139, # 24, p. 8267 - 8276
[19] Patent: JP2016/40292, 2016, A, . Location in patent: Paragraph 0038-0040
[20] Patent: KR101879905, 2018, B1, . Location in patent: Paragraph 0185-0189
[21] European Journal of Medicinal Chemistry, 2009, vol. 44, # 5, p. 1900 - 1912
[22] ChemCatChem, 2014, vol. 6, # 5, p. 1340 - 1348
[23] Patent: WO2016/615, 2016, A1, . Location in patent: Paragraph 00482
[24] Patent: CN104817489, 2017, B, . Location in patent: Paragraph 0056-0057
[25] Patent: CN108530336, 2018, A, . Location in patent: Paragraph 0077; 0079; 0083
With hydrogenchloride; sodium nitrite In water; acetone at -10 - 20℃; for 3 h;
A solution of NaNO2 (110.4 g, 1.6 mol, 8 eq) in water (200 mL) was added dropwise to a solution of 5-bromoindole (XI) (39.2 g, 0.2 mol, 1 eq) in acetone (1000 mL) stirred at −10→0° C., while adding NaNO2 the solution temperature was maintained below 20° C. An aqueous 2N HCl solution (480 mL) was added slowly to the solution with vigorously stirring while keeping the internal temperature between 0 and 20° C. The solution was further stirred at 20° C. for 3 h after the addition. The solution was concentrated under reduced pressure to remove acetone while keeping the temperature below 35° C. The solid was collected by filtration and transferred to a flask. Cold (−10° C.) DCM (200 mL) was added and stirred for 30 min at −5° C., the solids were filtered and dried under vacuum at 40° C. to get 5-bromo-1H-indazole-3-carbaldehyde (XII) (34.0 g, 151 mmol, 76percent yield) as a brown solid. ESIMS found for C8H5BrN2O m/z 225 (M+H).
Stage #1: With water; sodium nitrite In acetone at -10 - 20℃; Stage #2: With hydrogenchloride In water at 20℃; for 3 h; Stage #3: at -5℃; for 0.5 h;
Step 1 A solution of NaNO2 (110.4 g, 1.6 mol, 8 eq) in water (200 mL) was added dropwise to a solution of 5-bromoindole (I) (39.2 g, 0.2 mol, 1 eq) in acetone (1000 mL) stirred at -10→0° C., while adding NaNO2 the solution temperature was maintained below 20° C. An aqueous 2N HCl solution (480 mL) was added slowly to the solution with vigorously stirring while keeping the internal temperature between 0 and 20° C. The solution was further stirred at 20° C. for 3 h after the addition. The solution was concentrated under reduced pressure to remove acetone while keeping the temperature below 35° C. The solid was collected by filtration and transferred to a flask. Cold (-10° C.) DCM (200 mL) was added and stirred for 30 min at -5° C., the solids were filtered and dried under vacuum at 40° C. to get 5-bromo-1H-indazole-3-carbaldehyde (II) (34.0 g, 151 mmol, 76percent yield) as a brown solid. ESIMS found for C8H5BrN2O m/z 225 (M+H).
Reference:
[1] Journal of the American Chemical Society, 2008, vol. 130, # 48, p. 16162 - 16163
[2] European Journal of Medicinal Chemistry, 2013, vol. 64, p. 498 - 511
[3] Medicinal Chemistry Research, 2013, vol. 22, # 9, p. 4492 - 4504
72
[ 10075-50-0 ]
[ 773873-77-1 ]
Reference:
[1] Tetrahedron, 2016, vol. 72, # 19, p. 2376 - 2385
[2] Organic Letters, 2016, vol. 18, # 24, p. 6504 - 6507
[3] Chemical Papers, 2018, vol. 72, # 6, p. 1369 - 1378
Example 15 5-Bromo-3-(2,4-dichlorobenzoyl)-1-ethyl-1H-indole [0380] 1a) 5-Bromo-1-ethyl-1H-indole [0381] In 75 mL of anhydrous dimethylformamide, place 3.06 g (76.5 mmol) of sodium hydride in 60% suspension in mineral oil. Stir and heat the mixture to 80 C. Add progressively, using a spatula, 5.0 g (25.5 mmol) of 5-bromo-1H-indole. Continue the heating for 30 minutes (until no further hydrogen is evolved). Cool the mixture to ambient temperature and add 4 mL (51 mmol) of iodoethane. Heat the mixture again to 80 C. for 2 hours. Hydrolyse the reaction medium. Extract with dichloromethane, dry the organic phase over anhydrous sodium sulfate and evaporate. Purify the evaporation residue by chromatography on silica gel with elution by dichloromethane (Int. 49). [0382] Yield: 89% [0383] Yellow oil
With hydrogenchloride; sodium nitrite; In water; acetone; at -10 - 20℃; for 3h;
A solution of NaNO2 (110.4 g, 1.6 mol, 8 eq) in water (200 mL) was added dropwise to a solution of 5-bromoindole (XI) (39.2 g, 0.2 mol, 1 eq) in acetone (1000 mL) stirred at -10?0 C., while adding NaNO2 the solution temperature was maintained below 20 C. An aqueous 2N HCl solution (480 mL) was added slowly to the solution with vigorously stirring while keeping the internal temperature between 0 and 20 C. The solution was further stirred at 20 C. for 3 h after the addition. The solution was concentrated under reduced pressure to remove acetone while keeping the temperature below 35 C. The solid was collected by filtration and transferred to a flask. Cold (-10 C.) DCM (200 mL) was added and stirred for 30 min at -5 C., the solids were filtered and dried under vacuum at 40 C. to get 5-bromo-1H-indazole-3-carbaldehyde (XII) (34.0 g, 151 mmol, 76% yield) as a brown solid. ESIMS found for C8H5BrN2O m/z 225 (M+H).
With hydrogenchloride; sodium nitrite; In water; for 2.41667h;
5-Bromoindole (3.93 g, 20 mmol) is added to an aqueous sodium nitrite solution (13.80 g, 200 mmol in 400 ml H2O), followed slowly, over 25 min, by a 6N aqueous HCl solution (30 ml). The reaction mixture is vigorously stirred for 3 h and then filtered. The solid collected is taken up in AcOEt. This solution is dried and evaporated
With NaH; phosphoric acid; In tetrahydrofuran; N,N-dimethyl-formamide;
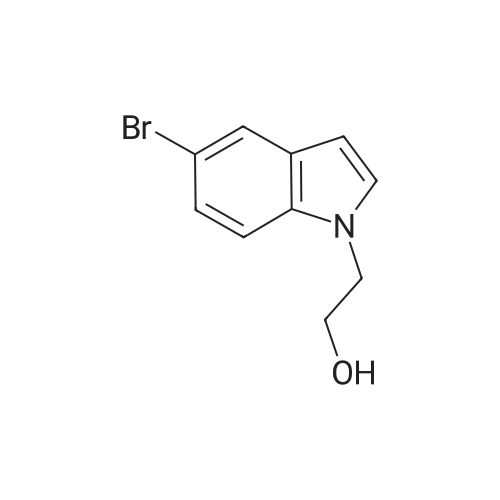
1-(2-Hydroxyethyl)-5-bromoindole: Under a N2 atmosphere, 2-bromoethanol (17.6 g, 141 mmol) and 2-methoxypropene (10.1 g, 141 mmol) were stirred in 71 mL of THF at 0 C. for 30 minutes The resulting solution was added to a stirring mixture of 5-bromoindole (22.83 g, 116 mmol) and 60% NaH (4.62 g, 193 mmol) in 40 mL of DMF and 60 mL of THF. The solution was stirred at ambient temperature for 4 hours. The reaction mixture was worked up by quenching the excess of NaH with water and removing the aqueous layer. The organic layer was vigorously stirred with 200 mL of 2% aqueous phosphoric acid for 5 hours when the layers were separated. The organic layer was washed with water (2*200 mL) and the solvent removed. The residue was purified by column chromatography (30:70 EtOAc-Hex) to give 17 g of 1-(2-hydroxylethyl)-5-bromoindole. Tris[1-(2-t-Butyldimethylsilyloxyethyl)indol-5-yl]bismuthane:
With NaH; phosphoric acid; In tetrahydrofuran; N,N-dimethyl-formamide;
1-(2-Hydroxyethyl)-5-bromoindole: Under a N2 atmosphere, 2-bromoethanol (17.6 g, 141 mmol) and 2-methoxypropene (10.1 g, 141 mmol) were stirred in 71 mL of THF at 0 C. for 30 minutes The resulting solution was added to a stirring mixture of 5-bromoindole (22.83 g, 116 mmol) and 60% NaH (4.62 g, 193 mmol) in 40 mL of DMF and 60 mL of THF. The solution was stirred at ambient temperature for 4 hours. The reaction mixture was worked up by quenching the excess of NaH with water and removing the aqueous layer. The organic layer was vigorously stirred with 200 mL of 2% aqueous phosphoric acid for 5 hours when the layers were separated. The organic layer was washed with water (2*200 mL) and the solvent removed. The residue was purified by column chromatography (30:70 EtOAc-Hex) to give 17 g of 1-(2-hydroxylethyl)-5-bromoindole.
STEP 7A 1-(2-Hydroxyethyl)-5-bromoindole A mixture of NaOH (4.4 gm, 0.011 mol) in DMSO (175 ml) was stirred at 100 C. for 5 hours at which time it was cooled to 20 C. To this mixture was added 5-bromoindole (20 gm, 0.102 mol) and the reaction was stirred for 8 hours at room temperature. A solution of ethylene oxide (5.1 gm, 0.125 mol) in DMSO (20 ml) was prepared by bubbling the gas into DMSO. To the bromoindole reaction mixture was slowly added the ethylene oxide solution and stirring was continued for another 2.5 hours. The reaction mixture was then poured into ice water and extracted twice with diethyl ether. The combined ether extracts were concentrated in vacuo whereupon crystallization took place. The crude product was recrystallized from diethyl ether:hexanes (3:2) to afford the title compound (6.25 gm).
With sodium hydroxide; In dimethyl sulfoxide;
STEP 1A 1-(2-Hydroxyethyl)-5-bromoindole A mixture of NaOH (4.4 gm, 0.011 tool) in DMSO (175 ml) was stirred at 100 C. for 5 hours at which time it was cooled to 20 C. To this mixture was added 5-bromoindole (20 gm, 0.102 mol) and the reaction was stirred for 8 hours at room temperature. A solution of ethylene oxide (5.1 gm, 0.125 mol) in DMSO (20 ml) was prepared by bubbling the gas into DMSO. To the bromoindole reaction mixture was slowly added the ethylene oxide solution and stirring was continued for another 2.5 hours. The reaction mixture was then poured into ice water and extracted twice with diethyl ether. The combined ether extracts were concentrated in vacuo whereupon crystallization took place. The crude product was recrystallized from diethyl ether:hexanes (3:2) to afford the title compound (6.25 gm).
With sodium hydroxide; In dimethyl sulfoxide;
STEP 25A 1-(2-Hydroxyethyl)-5-bromoindole A mixture of NaOH (4.4 gm, 0.011 mol) in DMSO (175 ml) was stirred at 100 C. for 5 hours at which time it was cooled to 20 C. To this mixture was added 5-bromoindole (20 gm, 0.102 mol) and the reaction was stirred for 8 hours at room temperature. A solution of ethylene oxide (5.1 gm, 0.125 mol) in DMSO (20 ml) was prepared by bubbling the gas into DMSO. To the bromoindole reaction mixture was slowly added the ethylene oxide solution and stirring was continued for another 2.5 hours. The reaction mixture was then poured into ice water and extracted twice with diethyl ether. The combined ether extracts were concentrated in vacuo whereupon crystallization took place. The crude product was recrystallized from diethyl ether:hexanes (3:2) to afford the title compound (6.25 gm).
With sodium hydroxide; In dimethyl sulfoxide;
STEP 25A 1-(2-Hydroxyethyl)-5-bromoindole A mixture of NaOH (4.4 gm, 0.011 mol) in DMSO (175 ml) was stirred at 100 C. for 5 hours at which time it was cooled to 20 C. To this mixture was added 5-bromoindole (20 gm, 0.102 mol) and the reaction was stirred for 8 hours at room temperature. A solution of ethylene oxide (5.1 gm, 0.125 mol) in DMSO (20 ml) was prepared by bubbling the gas into DMSO. To the bromoindole reaction mixture was slowly added the ethylene oxide solution and stirring was continued for another 2.5 hours. The reaction mixture was then poured into ice water and extracted twice with diethyl ether. The combined ether extracts were concentrated in vacuo whereupon crystallization took place. The crude product was recrystallized from diethyl ether:hexanes (3:2) to afford the title compound (6.25 gm).
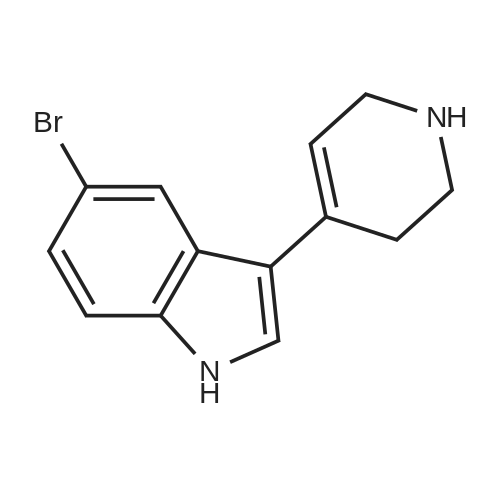
EXAMPLE 22 5-bromo-3-(1,2,3,6-tetrahydropyridin-4-yl)-1H-indole Beginning with 6.2 gm (31.6 mMol) 5-bromo-1H-indole and 4.7 gm (30.6 mMol) <strong>[40064-34-4]4-piperidone monohydrate hydrochloride</strong>, 7.93 gm (93%) of the title compound were recovered as a solid. m.p.=202-204 C. MS(FD): m/e=277.17 (M+)
EXAMPLE 22 5-bromo-3-(1,2,3,6-tetrahydropyridin-4-yl)-1H-indole Beginning with 6.2 gm (31.6 mMol) 5-bromo-1H-indole and 4.7 gm (30.6 mMol) <strong>[40064-34-4]4-piperidone monohydrate hydrochloride</strong>, 7.93 gm (93%) of the title compound were recovered as a solid. m.p.= 202-204C MS(FD): m/e=277.17 (M+)
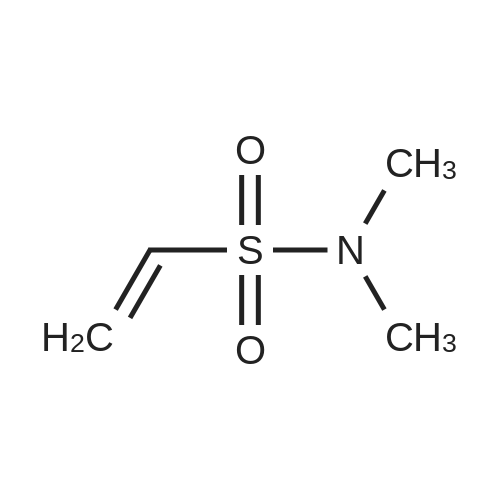
2-(1H-Indol-5-yl)-N,N-dimethylethenesulphonamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In acetonitrile;
(i) 2-(1H-Indol-5-yl)-<strong>[7700-07-4]N,N-dimethylethenesulphonamide</strong> A mixture of 5-bromoindole (7.7 g), <strong>[7700-07-4]N,N-dimethylethenesulphonamide</strong> (5.3 g) triethylamine (15 ml), acetonitrile (5 ml), palladium (II) acetate (0.35 g) and tri-o-tolylphosphine (0.95 g) was heated at 100 C. in an autoclave for 3 h. The resulting cooled mixture was partitioned between hydrochloric acid (2N, 300 ml) and ethyl acetate (2*150 ml). The combined extracts were dried (Na2 SO4) and evaporated in vacuo. The residue was purified by `flash` chromatography (V, 7 cm col.) to give the title compound as a crystalline solid (3.8 g) m.p. 148-150 C.
Add 5-bromoindole (0.500 g, 2.55 mmol) dropwise to a stirred slurry of sodium hydride (0.098 g, 4.08 mmol) in tetrahydrofuran. (12.75 mL) and stir for 1 h at room temperature. Add iodomethane (0.18 mL, 2.805 mmol) and let stir overnight. Pour over ice and dissolve further with water (100 mL) and extract into ethyl acetate (100 mL), wash with water (3 x 250 mL), saturated aqueous sodium chloride (2 x 250 mL), dry (sodium sulfate), filter, concentrate and purify (HPLC, eluting with 7: 3 hexanes:ethyl acetate) to give the title compound as a white solid (210 mg). HRMS m/z Calculated: 209.9901; Found: 209-9901
With copper(l) iodide; potassium carbonate; lithium chloride; In N,N-dimethyl-formamide; at 120℃; for 64h;Inert atmosphere;
Description 205-bromo-1-(3-fluoro-pyridin-4-yl)-1H-indole (D20)5-bromo-1H-indole [CA.S. 10075-50-0] (2 g, 10.202 mmol) was dissolved in DMF (16 ml). A nitrogen stream was bubbled through the mixture and then were added 3-Fluoro- 4-iodopyridine [CA.S. 22282-75-3] (2.502 g, 11.222 mmol), lithium chloride (0.432 g, 10.202 mmol), copper(I) iodide (0.0195 g, 0.102 mmol) and K2CO3 (4.23 g, 30.605 mmol). The reaction mixture was heated at 120 0C for 2 days. After cooling to room temperature, the reaction mixture was refilled with lithium chloride (0.100 g) and (copper(I) iodide (0.010) stirred at 1200C for 16 h. After cooling to room temperature, the reaction mixture was washed with NH3 (aqueous sat. solution) and extracted with DCM. The organic layer was separated, washed with water, dried (Na2SO4), and the solvent was evaporated in vacuo. The residue was purified by column chromatography (silica gel; eluent: Heptane/EtOAc up to 15percent as eluent). The desired fractions were collected and the solvent was evaporated in vacuo to yield intermediate compound D20 (1.37 g, 46 percent).
With potassium acetate; In water; at 100℃; for 24h;
General procedure: Typically, 1.0 ml H2O was added into the mixture of indole(1.0 mmol), aryl halide (1.2 mmol), KOAc (3.0 mmol), and the palladiumcatalyst (1.0 mol% Pd). The reaction mixture was stirred at 100 C for 24 h. After cooling to room temperature, most of the products are present in the aqueous phase. The suspension wascentrifuged. To completely collect the products residual remainingin the solid NU-1000, ethyl acetate was used to wash for threetimes (5 ml 3). The aqueous phase was extracted three timesusing ethyl acetate (5 ml 3). The two parts of organic phase werecombined and subsequently washed with water and brine andthen dried over Na2SO4. The product was purified by silica gelchromatography (mixture of light petroleum and ethyl acetate aseluent). The identification was conducted by 1H and 13C NMR measurement.
With aluminum (III) chloride; In 1,2-dichloro-ethane; at 80℃; for 7h;Inert atmosphere;
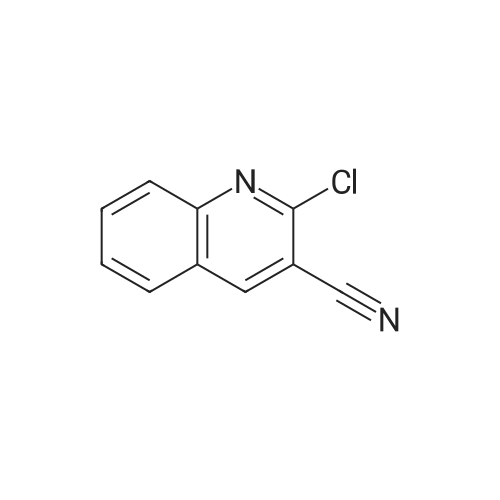
General procedure: A mixture of <strong>[95104-21-5]2-chloroquinoline-3-carbonitrile</strong> derivative10 (1, 1.0 equiv), an appropriate indole (2) (1.1 equiv) and anhydrous AlCl3 (1.2 equiv) in dichloroethane (5 mL) was stirred at 80 C for time indicated in Table 2 under a nitrogen atmosphere. After completion of the reaction, the mixture was poured into ice-cold water (15 mL), stirred for 10 min and then extracted with ethylacetate (3 × 20 mL). The organic layers were collected, combined, washed with cold water (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated under vacuum. The residue obtained was purified by column chromatography using ethylacetate /hexene to afford the desired product.
With tetra-(n-butyl)ammonium iodide; zinc trifluoromethanesulfonate; N-ethyl-N,N-diisopropylamine; In toluene; at 20℃;Inert atmosphere;
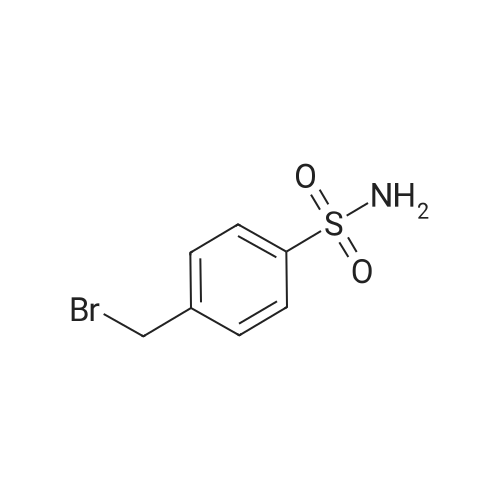
General procedure: Procedure A: To a solution of 1H-indole-5-sulfonamide/5-(methylsulfonyl)-1H-indole (2 equiv.), zinc triflate (1.2 equiv.) and tetrabutylammonium iodide (1 equiv.) in dry toluene (ca. 3 mL per mmol of indole) was added N,N-diisopropylethylamine (2.2 equiv.) under argon. The reaction mixture was heated at 50 °C for 30 min, followed by addition of the p-halo-benzyl bromide (1 equiv.). The mixture was stirred overnight at 50 °C under argon. The reaction was quenched with saturated aqueous NH4Cl, diluted with distilled water and extracted with ethyl acetate. The combined organic layers were washed with water and brine, dried over sodium sulfate anhydrous, filtered and concentrated. The samples were further purified with silica flash chromatography then PTLC.
With iodine; In 1,2-dichloro-ethane; at 80℃; for 17h;
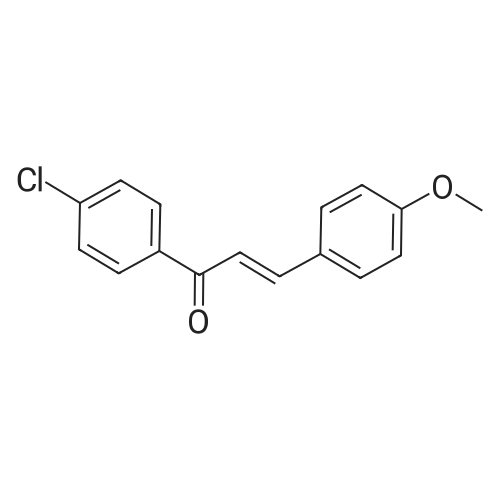
General procedure: To a mixture of piplartine (1 mmol) and Indole (3 mmol), Iodine (10 mol %) was added. The contents were refluxed in dichloroethane (5 mL) for an appropriate time (12-48 h) and reaction was monitored by thin-layer chromatography (TLC). After complete conversion, the solvent was evaporated, and the product was washed with hypo solution (10 mL), and then extracted with chloroform. The combined organic layers was dried over anhydrous sodium sulphate and evaporated under reduced pressure, purified by silica-gel column chromatography to afford pure product mono-adduct (2a-2k) and di-adduct (3a-3k).
With iodine; In 1,2-dichloro-ethane; for 36h;Reflux;
Experimental Procedure for (2g & 3g); To a mixture of piplartine (0.317 g, 1 mmol) and 5-bromoindole (0.585 g, 3 mmol), Iodine (0.0127 g, 10 mol %) was added. The contents were refluxed in 1,2-dichloroethane (5 ml) for an appropriate time (36 h). The reaction was monitored by thin-layer chromatography (TLC). After complete conversion, the solvent was evaporated, and the product was washed with saturated hypo solution (10 ml), and then extracted with chloroform. The combined organic layer was dried over anhydrous sodium sulphate and evaporated using rotary evaporator, purified by silica-gel (60-120 mesh) column chromatography by Ethylacetate:Hexane (3:7) to afford pure product mono adduct (2g) and di adduct (3g).
With lithium tert-butoxide; In N,N-dimethyl-formamide; at 100℃; under 760.051 Torr; for 24h;
General procedure: In a dried two-necked test tube was charged with LiOtBu (160 mg, 2.00 mmol) and indole 1a (23.4 mg, 0.4 mmol). The reaction vessel was evacuated under high vacuum and the atmosphere was replace with a balloon of CO2. Then DMF (2 mL) was added and the mixture was stirred for 24 h at 100C. Then the result mixture was cooled and carefully quenched with a solution of HCl (2 N) and extracted with EtOAc (5x). The combined organic layers were washed with water (2x), brine (1x) and dry over MgSO4. The dried organics were concentrated under reduce pressure and the residue was purified by preparative TLC (hexane:acetone = 1:1) to afford the desired product 2a (153.0 mg, 95%) as a white solid.
With copper diacetate; potassium carbonate; copper(I) bromide; sodium hydroxide; In N,N-dimethyl-formamide; at 100 - 110℃; for 16.1667h;
5-Bromo-1-(2-methylpyrimidin-5-yl)-1H-indole (13A) 5-Bromo-2-methylpyrimidine (0.971 g, 5.61 mmol) was added to a solution of 5-bromo-1H-indole (1 g, 5.1 mmol) and copper(I) bromide (0.073 g, 0.51 mmol) in DMF (10 mL) followed by potassium carbonate (1.762 g, 12.75 mmol), and the resulting mixture was stirred at 100° C. for 10 min. NaOH (0.153 g, 3.83 mmol) and copper(II) acetate (0.009 g, 0.051 mmol) were added at 110° C., and the reaction mixture was stirred for 16 h. Insoluble solids were filtered, and the filtrate was concentrated. The residue was partitioned between ethyl acetate and water. Separated organic layer was dried over sodium sulphate and filtered. The filtrate was concentrated in vacuo and purified by flash chromatography using 10percent ethyl acetate in hexane to afford the title compound (0.25 g, 16percent) as an off-white solid. 1H NMR (300 MHz, CDCl3): delta 8.81 (s, 2H), 7.83 (d, J=1.5 Hz, 1H), 7.36 (dd, J1=1.8 Hz, J2=8.7 Hz, 1H), 7.33-7.26 (m, 2H), 6.71 (d, J=3.3 Hz, 1H), 2.82 (s, 3H). ESI-MS m/z=288 (M+H)+.
16%
5-Bromo-2-methyl pyrimidine (0.971 g, 5.61 mmol) was added to a solution of 5-bromo- lH-indole (1 g, 5.1 mmol) and copper(I) bromide (0.073 g, 0.51 mmol) in DMF (10 mL) followed by potassium carbonate (1.762 g, 12.75 mmol), and the resulting mixture was stirred at 100 °C for 10 min. NaOH (0.153 g, 3.83 mmol) and copper(II) acetate (0.009 g, 0.051 mmol) were added at 1 10 °C, and the reaction mixture was stirred for 16 h. Insoluble solids were filtered, and the filtrate was concentrated. The residue was partitioned between ethyl acetate and water. Separated organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated in vacuo and purified by flash chromatography using 10percent ethyl acetate in hexanes to afford the title compound (0.25 g, 16percent) as an off-white solid. H NMR (300 MHz, CDC13):8 8.81 (s, 2H), 7.83 (d, J= 1.5 Hz, 1H), 7.36 (dd, J; = 1.8 Hz, J2 = 8.7 Hz, 1H), 7.33 - 7.26 (m, 2H), 6.71 (d, J= 3.3 Hz, 1H), 2.82 (s, 3H). ESI-MS m/z = 288 (M+H)+.
l-Iodo-3-(trifluoromethyl)benzene (2.204 g, 7.65 mmol) was added to a solution of 5- bromo- lH-indole (1.5 g, 7.65 mmol) and copper(I) bromide (0.110 g, 0.765 mmol) in DMF (30 mL) followed by potassium carbonate (2.1 15 g, 15.30 mmol) and the mixture was stirred at 100 C for 10 min. NaOH (0.0131 g, 0.765 mmol) and copper(II) acetate (0.138 g, 0.765 mmol) were then added at 1 10 C, and the reaction mixture was stirred for 16 h. Insoluble solids were filtered, the filtrate was concentrated, and the residue was partitioned between ethyl acetate and water. The separated organic layer was dried over sodium sulfate and filtered, and the filtrate was concentrated in vacuo to afford the title compound (0.8 g, 29%), which was carried on to the next step without any further purification. ESI-MS m/z = 356 (M+H)+.
With copper diacetate; potassium carbonate; copper(I) bromide; sodium hydroxide; In N,N-dimethyl-formamide; at 100 - 110℃; for 16.1667h;
5-Bromo-1-(3-(trifluoromethoxy)phenyl)-1H-indole (2A) 1-Iodo-3-(trifluoromethyl)benzene (2.204 g, 7.65 mmol) was added to a solution of 5-bromo-1H-indole (1.5 g, 7.65 mmol) and copper(I) bromide (0.110 g, 0.765 mmol) in DMF (30 mL) followed by potassium carbonate (2.115 g, 15.30 mmol) and the mixture was stirred at 100 C. for 10 min. NaOH (0.0131 g, 0.765 mmol) and copper(II) acetate (0.138 g, 0.765 mmol) were then added at 110 C., and the reaction mixture was stirred for 16 h. Insoluble solids were filtered, the filtrate was concentrated, and the residue was partitioned between ethyl acetate and water. The separated organic layer was dried over sodium sulphate and filtered, and the filtrate was concentrated in vacuo to afford the title compound (0.8 g, 29%), which was carried on to the next step without any further purification. ESI-MS m/z=356 (M+H)+.
5-Bromo-lH-indole (1.9 g, 9.69 mmol) was added to a solution of 2-(5-bromopyridin-2- yl)propan-2-ol (2.304 g, 10.66 mmol) and copper(I) bromide (0.139 g, 0.97 mmol) in DMF (35 mL) followed by potassium carbonate (3.35 g, 24.23 mmol), and the resulting mixture was stirred at 100 C for 10 min. NaOH (0.291 g, 7.27 mmol) and copper(II) acetate (0.018 g, 0.097 mmol) were added at 130 C, and the reaction mixture was stirred for 12 h. Insoluble solids were filtered, the filtrate was concentrated, and the residue was partitioned between ethyl acetate and water. Separated organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated in vacuo and purified by flash chromatography using 15% ethyl acetate in hexanes to afford the title compound (0.6 g, 15.89%) as a white solid. H NMR (300 MHz, DMSO-i/6): delta 8.74 (d, J= 2.7 Hz, 1H), 8.04 (dd, J = 2.7 Hz, J2 = 8.4 Hz, 1H), 7.88 - 7.84 (m, 2H), 7.78 (d, J= 3.3 Hz, 1H), 7.5 (d, J= 9.0 Hz, 1H), 7.33 (dd, Ji = 1.8 Hz, J2 = 8.7 Hz, 1H), 6.74 (d, J= 3.3 Hz, 1H), 5.35 (s, 1H), 1.5 (s, 6H). ESI-MS m/z = 331 (M+H)+.
With caesium carbonate; In N,N-dimethyl-formamide; at 70℃; for 1h;
Oxetan-3-yl methanesulfonate (1.397 g, 9.18 mmol) was added to a solution of 5-bromo- lH-indole (1.5 g, 7.65 mmol) in DMF (15 mL) followed by cesium carbonate (3.74 g, 1 1.48 mmol), and the mixture was stirred at 70 C for 1 h. Insoluble solids were filtered, the filtrate was concentrated and the residue was partitioned between ethyl acetate and water. Separated organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated under reduced pressure and purified by flash chromatography using 5% ethyl acetate in hexanes to afford the title compound (0.6 g, 30.5%) as an oil. lH NMR (400 MHz, CDC13): delta 7.77 (d, J= 1.6 Hz, 1H), 7.42 (d, J= 3.2 Hz, 1H), 7.35 (d, J= 8.8 Hz, 1H), 7.30 (dd, J = 2.0 Hz, J2 = 8.8 Hz, 1H), 6.54 (d, J= 2.8 Hz, 1H), 5.58 - 5.45 (m, 1H), 5.17 (t, J= 7.2 Hz, 2H), 5.05 (t, J= 6.8 Hz, 2H). ESI-MS m/z = 252 (M+H)+.
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,4-dioxane; water; at 80℃; for 0.5h;Inert atmosphere; Microwave irradiation;
General procedure: A suspension of 9 (200 mg, 1.02 mmol), quinolin-3-ylboronic acid (211 mg, 1.22 mmol), Pd(Ph3)4 (118 mg, 0.10 mmol) and K2CO3 (422 mg, 3.06 mmol) in 1,4-dioxane (8 mL) and H2O (2 mL) was degassed with argon for 10 min and then heated in a microwave reactor at 80 C for 30 min. The mixture was cooled to room temperature and partitioned between EtOAc (50 mL) and sat. aq. Na2CO3 (50 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2SO4 and the solvent evaporated under reduced pressure. The resultant residue was purified by silica gel flash-column chromatography, with CH2Cl2/EtOAc (8:2) as the eluent, to yield the title compound as a white solid (170 mg, 68%)
Step 1 A solution of NaNO2 (110.4 g, 1.6 mol, 8 eq) in water (200 mL) was added dropwise to a solution of 5-bromoindole (I) (39.2 g, 0.2 mol, 1 eq) in acetone (1000 mL) stirred at -10?0 C., while adding NaNO2 the solution temperature was maintained below 20 C. An aqueous 2N HCl solution (480 mL) was added slowly to the solution with vigorously stirring while keeping the internal temperature between 0 and 20 C. The solution was further stirred at 20 C. for 3 h after the addition. The solution was concentrated under reduced pressure to remove acetone while keeping the temperature below 35 C. The solid was collected by filtration and transferred to a flask. Cold (-10 C.) DCM (200 mL) was added and stirred for 30 min at -5 C., the solids were filtered and dried under vacuum at 40 C. to get 5-bromo-1H-indazole-3-carbaldehyde (II) (34.0 g, 151 mmol, 76% yield) as a brown solid. ESIMS found for C8H5BrN2O m/z 225 (M+H).
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,4-dioxane; water; at 80℃; for 0.5h;Inert atmosphere; Microwave irradiation;
General procedure: A suspension of 9 (200 mg, 1.02 mmol), quinolin-3-ylboronic acid (211 mg, 1.22 mmol), Pd(Ph3)4 (118 mg, 0.10 mmol) and K2CO3 (422 mg, 3.06 mmol) in 1,4-dioxane (8 mL) and H2O (2 mL) was degassed with argon for 10 min and then heated in a microwave reactor at 80 C for 30 min. The mixture was cooled to room temperature and partitioned between EtOAc (50 mL) and sat. aq. Na2CO3 (50 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2SO4 and the solvent evaporated under reduced pressure. The resultant residue was purified by silica gel flash-column chromatography, with CH2Cl2/EtOAc (8:2) as the eluent, to yield the title compound as a white solid (170 mg, 68%)
3-[2-(1H-indol-5-yl)ethynyl]benzoic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Example 75: Synthesis of 3-[2-(l,3-thiazol-4-yl)ethynyl]benzoic acid: 3-[2-(l,3-thiazol-4-yl)ethynyl]benzoic acid. A mixture of 3-ethynyl-l-yl- benzoic acid methyl ester (40 mg, 0.25 mmol), 4-bromo-thiazole (82 mg, 0.5 mmol), palladium tetrakis-triphenylphosphine (29 mg, 0.025 mmol) and copper iodide (9.5 mg, 0.05 mmol), potassium carbonate (69 mg, 0.5 mmol) in 1,2- dimethoxyethane/water (1 mL/0.3 mL) was degassed with N2 for 5 minutes and then heated at 60 C for 4 hours. After cooling to ambient temperature, the crude mixture was filtered through celite and washed with dichloromethane. The filtrate was concentrated and purified by preparative thin layered chromatography eluting with ethyl acetate/hexane (30%) to give the ester intermediate. To this intermediate in tetrahydrofuran/methanol (1 mL/0.2 mL) was added sodium hydroxide solution (2 N in water, 0.2 mL, 0.4 mmol) and the solution was stirred at room temperature for 18 hours. 1 N hydrochloric acid aqueous solution was added dropwise until pH = 1 and the reaction mixture was purified through preparative HPLC to give 18 mg (22% for 2 steps) of the pure product as a white solid. FontWeight="Bold" FontSize="10" H NMR (300MHz, DMSO) delta = 13.2 (br. s., 1H), 9.18 (d, J=2.1 Hz, 1H), 8.19 (d, J=1.8 Hz, 1H), 8.05 (t, J=1.5 Hz, 1H), 7.98 (td, J=1.4, 7.8 Hz, 1H), 7.81 (td, J=1.4, 7.8 Hz, 1H), 7.57 (t, J=7.8 Hz, 1H). LCMS (ESI) mlz 230.0 (M+l)+. Example 77: Synthesis of 3-[2-(lH-indol-5-yl)ethynyl]benzoic acid: 3-[2-(lH-indol-5-yl)ethynyl]benzoic acid was prepared by the same procedure as example 75. NMR (300MHz, CDC13) delta = 9.30 (br. s., 1H), 8.23 (t, J=1.8 Hz, 1H), 7.98 (td, J=1.5, 7.9 Hz, 1H), 7.87 (d, J=0.9 Hz, 1H), 7.72 (td, J=1.3, 7.9 Hz, 1H), 7.47 - 7.23 (m, 4H), 6.59 - 6.50 (m, 1H). LCMS (ESI) mlz 262.0 (M+l)+.
5-(1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazol-4-yl)-1H-indole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
9 g
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; caesium carbonate; In water; N,N-dimethyl-formamide; at 90℃; for 12h;Inert atmosphere;
Reaction Step 1 Synthesis of 5-(1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazol-4-yl)-1H-indole To a solution of 5-bromo-1H-indole (10.0 g, 51.0 mmol, 1.0 eq) in a mixture of DMF and water (9:1, 100 mL), 1-(tetrahydro-2H-pyran-2-yl)-4-(4,4,5,5-tetramethyl-1,3-dioxolan-2-yl)-1H-pyrazole (21.2 g, 76.5 mmol, 1.5 eq) was added at room temperature, followed by Cs2CO3 (33.0 g, 102 mmol, 2.0 eq). The mixture was purged with argon for 15 min, Pd(dppf)2Cl2.DCM (4.1 g, 5.1 mmol, 0.1 eq) was added followed by purging with argon for further 10 min and the mixture was heated at 90 C. for 12 h. After completion of the reaction (monitored by TLC 30% ethyl acetate in hexanes Rf=0.35), the mixture was cooled to room temperature and filtered through a bed of diatomaceous earth, washing with ethyl acetate. The combined filtrate was washed with water followed by brine, the organic layer was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (100-200 mesh), eluting with a 20-30% gradient of ethyl acetate in hexanes to afford 5-(1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazol-4-yl)-1H-indole (9.0 g, 66%) as a white solid. LCMS purity: 68%; (ES+): m/z 268.06 (M+H+); tr=1.85 min.
With norborn-2-ene; dichloro bis(acetonitrile) palladium(II); potassium carbonate; In N,N-dimethyl acetamide; water; at 70℃;
General procedure: A high pressure tube equipped with a magnetic stirring bar was charged with indole substrate (0.2mmol, 1 equiv.), norbornene (0.4mmol, 2 equiv.), the base (0.4mmol, 2 equiv. K2CO3 or 2 equiv. KHCO3, as indicated),, and PdCl2(MeCN)2 (0.02mmol, 10 mol %). A solution of water (0.5 M) in DMA (1mL) was added via syringe, and then the aryl iodide (0.4mmol, 2 equiv.) was added via syringe. The reaction mixture was then placed in a preheated oil bath at 70 C (or 90 C, as indicated). Vigorous stirring was applied. The reaction was monitored by TLC. Upon completion, the reaction mixture was cooled to room temperature, diluted with EtOAc, and filtered. The filtrate was extracted with H2O (3 times) and brine, the organic phase was concentrated in vacuum to remove the solvent. The residue was directly submitted to flash column chromatography to afford the 2-arylindole product. The spectral data of the products were in accordance with those reported in the literature1.
48%
With potassium acetate; palladium diacetate; triphenylphosphine; In water; at 110℃; for 24h;Inert atmosphere;
With potassium acetate; palladium diacetate; triphenylphosphine; In water; at 110℃; for 24h;Inert atmosphere;
Under a nitrogen stream , 5-bromo-1H-indole (25g, 0.13mol), Iodobenzene (31.22g, 0.15mol), Pd(OAc)2 (1.43g, 5.mol%), Triphenylphosphine (1.67g.5mol%), KOAc (37.55g, 0. Were mixed 38 mole ) and H2O (300ml), and stirred for 24 hours at 110 C. After completion of the reaction, and extracted with ethyl acetate , to remove water with MgSO4 , Karamukuro Chromatograph By (hexane :: EA = 10 1 (v / v 5-bromo-2-phenyl-1H-indole (16.66g, 48% yield) .
48%
With potassium acetate; palladium diacetate; triphenylphosphine; In water; at 110℃; for 24h;Inert atmosphere;
Under a nitrogen stream, 5-bromo-1H-indole (25 g, 0.13 mmol), iodobenzene (31.22g, 0.15mol), Pd (OAc) 2 (1.43g, 5mol%), triphenylphosphine (1 .67g, 5mol%), KOAc (37.55g, 0.38mol), and H2O to (300 ml) were mixed and stirred for 24 hours at 110 C..After completion of the reaction, and extracted with ethyl acetate, water was removed by MgSO4, column chromatography (hexane: EA = 10: 1 (v / v)) and purified by 5-bromo-2-phenyl -1H- indole ( 16.66g, 48% yield).
48%
With potassium acetate; palladium diacetate; triphenylphosphine; In water; at 110℃; for 24h;Inert atmosphere;
Under a stream of nitrogen, 5-bromo-1H-indole (25 g, 0.13 mol), Iodobenzene (31.22 g, 0.15 mol), Pd(OAc)2 (1.43 g, 5 mol%), Triphenylphosphine (1.67 g, 5 mol%), mixture of KOAc (37.55 g, 0.38 mol) and H2O (300 ml) and stirred for 24 hours at 110C. After the reaction was terminated to remove water in the following MgSO4 extracted with ethyl acetate and purified by column chromatography (Hexane: EA = 10: 1 (v / v)) to give 5-bromo-2-phenyl-1H-indole (16.66 g, a yield of 48%).
48%
With potassium acetate; palladium diacetate; triphenylphosphine; In water; at 110℃; for 24h;Inert atmosphere;
Under a nitrogen stream, 5-bromo -1H- indole (25 g, 0.13 mol), iodobenzene (31.22g, 0.15mol), Pd (OAc) 2 (1.43g, 5mol%), triphenylphosphine (1 .67g, 5mol%), KOAc (37.55g, 0.38mol), and H2O to (300 ml) were mixed and stirred for 24 hours at 110 C.. After completion of the reaction, and extracted with ethyl acetate, to remove water with MgSO4, purified by column chromatography (hexane: EA = 10: 1 (v / v)) to give the 5-bromo-2-phenyl -1H- indole It was obtained (16.66g, 48% yield).
48%
With potassium acetate; palladium diacetate; triphenylphosphine; In water; at 110℃; for 24h;Inert atmosphere;
5-bromo-1H-indole (25 g, 0.13 mol) was added under a nitrogen stream, Iodobenzene (31.22 g, 0.15 mol), Pd (OAc) 2 (1.43 g, 5 mol%),Triphenylphosphine (1.67 g, 5 mol%), KOAc (37.55 g, 0.38 mol) andH 2 O (300 ml) were mixed and stirred at 110 C for 24 hours.After the reaction was completed, the reaction mixture was extracted with ethyl acetate, the water was removed with MgSO4,Purification by column chromatography (Hexane: EA = 10: 1 (v / v)) gave 5-bromo-2-phenyl-1H-indole (16.66 g, yield 48%).
5-bromo-3-(7-bromo-3,4-dihydronaphthalen-2-yl)-1H-indole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
70%
With L-Tartaric acid; 1,1-Dimethylurea; at 70℃; for 3h;
General procedure: In a typical experiment, 1.5 g of L-(+)-tartaric acid - N,N?-dimethylurea (30:70) mixture was heated at 70 C to obtain a clear melt. To this melt, 1.2 mmol of indole and 1.0 mmol of ketone were added at 70 C. At the conclusion (TLC monitoring by mini work up), the reaction mixture was quenched by adding water while still hot. The reaction mixture was cooled to room temperature and the solid product was filtered and washed with water (2 5 mL). The crude product was dried under vacuum and then purified by silica gel column chromatography.
With toluene-4-sulfonic acid; In acetonitrile; at 50.0℃; for 72.0h;
General procedure: A solution of indoles or pyrroles 1 (1.0 mmol), tanshinones 2 (0.40 mmol) and 20 mol% TsOH (0.08 mmol) in CH3CN (5.0 mL) was stirred at 50 oC for 72 h. After completion of the reaction, as indicated by TLC, the removal of solvent and purification by flash column chromatography (hexane/EtOAc = 8:1~5:1) were carried out to furnish the corresponding products 3.
With bis-triphenylphosphine-palladium(II) chloride; potassium phosphate; In 1,4-dioxane; water; at 95℃;Inert atmosphere;
General procedure: The halo aryl (1.0 equiv) was dissolved in a mixture of water:dioxane (1:1). The boronic acid or ester(1.5 equiv) and potassium phosphate (5.0 equiv) were added. The solution was degassed byvacuum/argon cycles (10 times) before addition of PdCl2(PPh3)2 (10 mol%) and further degassed (5times). The resulting mixture was stirred at 95 C under argon atmosphere for 16-20 hours. Thereaction mixture was filtered through Celite and diluted with water (approx. 30 mL) before washingwith chloroform (3 x 30 mL). If not stated otherwise, the aqueous phase was concentrated underreduced pressure and applied to a C18 precolumn before purification on a 10g or 60 g C18 column witha gradient of acetonitrile in water (10-100%) to yield the desired product.
2-(5-bromo-1H-indol-3-ylthio)-6-(difluoromethoxy)-1H-benzo[d]imidazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
81%
With manganese(III) triacetate dihydrate; acetic acid; at 80℃; for 2h;Inert atmosphere;
General procedure: Manganese triacetate (2 mmol) is added portion wise to a pre dissolved solution of Indole (1mmol) and benzimidazolethiol (1mmol) in acetic acid (10mL) at room temperature under Nitrogen atmosphere. The reaction mixture is stirred at 80 C for 2 hours. After completion of reaction, as monitored by T.L.C analysis, the reaction mixture was quenched by the addition of water 20 ml. The organic compounds are extracted with ethyl acetate (3x20ml). The combined layers are dried over anh.Na2SO4. The required product is purified by column chromatography, eluted with 8% methanol in DCM to get the product. The product is confirmed by 1H, 13C, NMR, IR and mass
2-(5-bromo-1H-indol-3-ylthio)-6-methoxy-1H-benzo[d]imidazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
With manganese(III) triacetate dihydrate; acetic acid; at 80℃; for 2h;Inert atmosphere;
General procedure: Manganese triacetate (2 mmol) is added portion wise to a pre dissolved solution of Indole (1mmol) and benzimidazolethiol (1mmol) in acetic acid (10mL) at room temperature under Nitrogen atmosphere. The reaction mixture is stirred at 80 C for 2 hours. After completion of reaction, as monitored by T.L.C analysis, the reaction mixture was quenched by the addition of water 20 ml. The organic compounds are extracted with ethyl acetate (3x20ml). The combined layers are dried over anh.Na2SO4. The required product is purified by column chromatography, eluted with 8% methanol in DCM to get the product. The product is confirmed by 1H, 13C, NMR, IR and mass
General procedure: A mixture of acetylacetone (1 mmol) and arylglyoxal (1 mmol) in water (15mL) was stirred at 60 oC temperature for 20 min. Indole (1 mmol) was added and the mixture was stirred for 5 min at 60 oC. Then aliphatic amine (1 mmol) was added to this mixture. The reaction mixture was then stirred at 60 oC temperature for 1 h. The solid was filtered off and washed with diethyl ether (20 mL) to afford the pure product.
5-bromo-1H-indole (700 mg, 3.57 mmol) was dissolved in anhydrous DMF (10 mL), NaH (60%, 429 mg, 10.71mmol) was added at 0 C, the reaction solution was slowly warmed to room temperature, and allowed to proceedovernight. Then TDI01286-2 (579 mg, 4.29 mmol) was added at room temperature, and the reaction was further stirredfor 3 hours at room temperature. LC-MS indicated the reaction was complete. The reaction solution was slowly addedto 100 mL water, and stirred at room temperature for 30 min. A large amount of solid precipitated, and was filtered bysuction. The filter cake was washed, collected and dried to afford TDI01286-3 (800 mg, crude product).1H NMR (400 MHz, DMSO-d6) delta 8.17 (d, J = 7.2 Hz, 1H), 7.61 (s, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.33 (d, J = 3.2 Hz, 1H),7.15 (d, J = 8.4 Hz, 1H), 6.46 (d, J = 2.8 Hz, 1H), 4.77 (s, 2H), 3.88-3.80 (m, 1H), 1.09 (d, J = 6.4 Hz, 6H). MS m/z (ESI):297.2 [M+H].
4-bromo-2-(5-chloroquinolin-3-yl)aniline[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
56%
With tin(II) chloride dihdyrate; In ethanol; at 80℃; for 0.5h;Microwave irradiation;
General procedure: 2-nitrobenzaldehyde (0.5 mmol), indole (0.5 mmol), and SnCl2·2H2O (3.0 equiv) in EtOH (3 mL) was taken in 50 mL round bottom flask. The resulting mixture was placed in microwave reactor (NmuTeCH) at 200 W and the temperature was set to 80 C for 30 min (and for conventional heating, the mixture was heated at 80 C for a period of the time till the completion of the reaction). The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature and then added NaHCO3 (1 M) solution till pH 8. The mixture was extracted with EtOAc. The combined organic layers were then dried with anhydrous Na2SO4. The organic layer was concentrated under reduced pressure and the residue was purified by column chromatography on silica gel (Hexane/EtOAc) to get a final product. All the synthesized compounds were characterized by mp, IR, NMR and Mass.
With potassium tert-butylate; In N,N-dimethyl-formamide; at 20℃; for 0.116667h;Microwave irradiation;
5-bromoindole (100 mg, 0.62 mmol),DMF (4 mL),Potassium tert-butoxide (171, 1.85 mmol, 3 eq.) was added to the reaction flask in turn.After stirring at room temperature for 5 minutes,After the potassium tert-butoxide is uniformly dispersed,Slowly inject into the reaction flaskEthyl trifluoroacetate (0.29 mL, 2.47 mmol, 4 eq.).400 W microwave irradiation for 2 minutes.After cooling to room temperature, extract twice with ethyl acetate and distilled water.Wash with saturated NaCl solution,Combine the organic phase,Drying the organic phase with anhydrous magnesium sulfate,Remove magnesium sulfate by filtration,Rotate the solvent,Column chromatography purification,Light green liquidN-ethyl-5-bromoindole(62 mg, yield 53%).
With water; sodium nitrite; In water; at -10 - 20℃; for 3h;
A solution of NaNO2 (110.4 g, 1.6 mol, 8 eq) in water (200 mL) was added dropwise to a solution of 5-bromoindole (XXVI) (39.2 g, 0.2 mol, 1 eq) in acetone (1000 mL) stirred at -100oC, while adding NaNO2 the solution temperature was maintained below 20oC. An aqueous 2N HCl solution (480 mL) was added slowly to the solution with vigorously stirring while keeping the internal temperature between 0 and 20oC. The solution was further stirred at 20oC for 3 h after the addition. The solution was concentrated under reduced pressure to remove acetone while keeping the temperature below 35oC. The solid was collected by filtration and transferred to a flask. Cold (-10oC) DCM (200 mL) was added and stirred for 30 min at -5oC, the solids were filtered and dried under vacuum at 40oC to get 5-bromo-1H-indazole-3-carbaldehyde (XXVII) (34.0 g, 151 mmol, 76% yield) as a brown solid. ESIMS found for C8H5BrN2O m/z 225 (M+H).
With potassium carbonate; In ethanol; at 140℃;Schlenk technique;
General procedure: A mixture of indole-3-carboxylic acid 1 (0.50 mmol) and K2CO3 (13.8 mg, 0.10 mmol, 20 mol%) in EtOH (3 mL) was added into a Schlenk flask (25 mL) and stirred at 140 C. The reaction was monitored by thin layer chromatography until the disappearance of starting material 1. Then the solvent was evaporated under reduced pressure and the residue was purified by column chromatography (petroleum ether/ethyl acetate 20:1 to 10:1).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






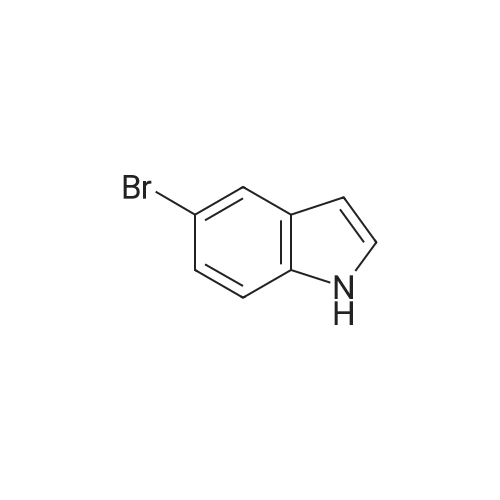

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping