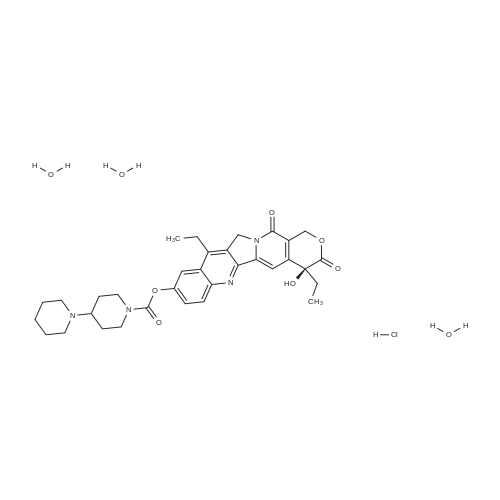
| 91.7% |
With hydrogenchloride; In water; acetone; at 22℃; for 25 - 46h;Product distribution / selectivity; |
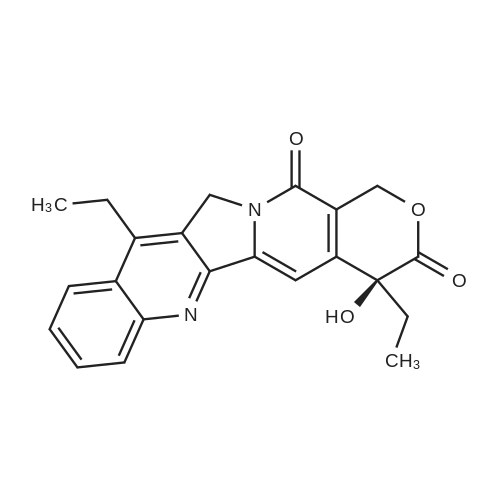
Irinotecan was suspended in acetonitrile or acetone in accordance with the amounts shown in Table 1 and dissolved by adding, 6 mol/L hydrochloric acid. To each mixture solution, 1 mg of c-type crystals separately prepared was added and the solution was stirred at 22C for 25 to 46 hours. The formed crystals were obtained by filtration, dried under reduced pressure, and subjected to a moisture absorption process performed by a saturated aqueous sodium chloride solution method until the crystals showed constant weight (about 80 hours). In this manner, the crystals of irinotecan hydrochloride were obtained. Table 1 shows the results of the preparations. [Table 1] No.Irinotecan (g)Solvent1) Hydrochloric acid2) Seed crystalsCrystallization time (hour)Yield (%)AcetonitrileAcetone 15.0801.0-4683.425.01201.0c form2580.5310.01201.05c form4176.341.03601.05c form4191.71) mL/g (Irinotecan)2) Times by mole relative to irinotecan The crystals of irinotecan hydrochloride obtained above were subjected to infrared absorption spectrum analysis, powder X-ray diffraction analysis, and thermoanalysis. The results are shown in Figures 1 to 3. In the infrared absorption spectrum, strong absorption was observed at wavelengths of about 1757 cm-1, 1712 cm-1, 1667 cm-1 (Figure 1). In the powder X-ray diffraction, diffraction peaks (2theta) were observed at 9.15, 10.00, 11.80, 12.20, 13.00 and 13.40; however no strong peak was observed at 11.00, which is intrinsic to the b-type crystals (Figure 2). In the thermoanalysis (differential scanning calorimetry), an endothermic peak near 90C due to dehydration was not observed, which is intrinsically observed in the b-type crystals (Figure 3). The moisture content of the crystals was measured by the Karl Fischer method. As a result, the moisture content of No.3 was 3.96%. Therefore, it turned out that the crystals above were obtained in the form of sesquihydrates (calculation value: 4.15%). From the results above, it was confirmed that the crystals of irinotecan hydrochloride prepared in accordance with Nos. 1 to 4 are c-type, which are different from the b-type crystals conventionally crystallized from water. |
| 91% |
With hydrogenchloride; In water; at 20℃; for 12h; |
0.63 g of irinotecan free base was placed in a 250 mL three-necked flask.Add 100 ml of water, add 12 ml of concentrated hydrochloric acid under stirring, stir at room temperature for 10 hours, and filter.The filtrate was extracted three times with methylene chloride, and the aqueous solution was concentrated to 30 ml under reduced pressure.The ice salt bath was cooled to a temperature of 5 C, and the crystals were filtered off and washed three times with water.Vacuum drying at room temperature (adding phosphorus pentoxide),The irinotecan hydrochloride trihydrate compound was obtained in an amount of 0.85 g, and the yield was 91%. |
| 90% |
With hydrogenchloride; In water; |
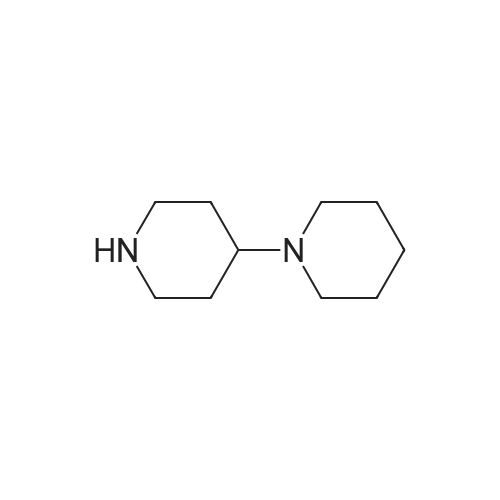
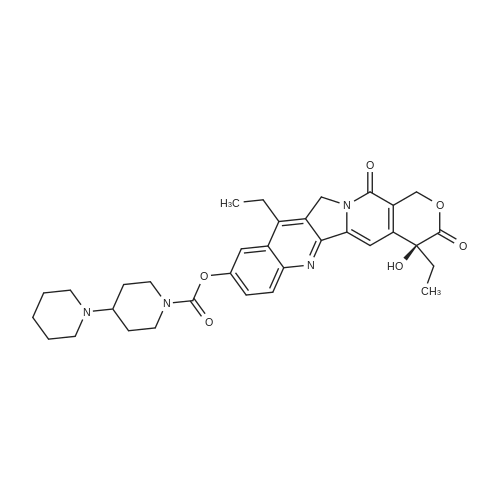
Example 3 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxy-camptothecin (I) In a suitable vessel were charged 7-Ethyl-10-hydroxycamptothecin (1 g), methylene dichloride, dimethylformamide (0.186 gm) and pyridine (3 ml) under nitrogen atmosphere. A solution of [1,4']bipiperidinyl-1'-carbonyl chloride (0.88 g), methylene dichloride and triethylamine (1 ml) was prepared and added to the above suspension and stirred at 30-40 C. for 2 hours. The solvent was distilled out under reduced pressure at 50 C. and hexane was added under stirring as an antisolvent to isolate crystalline compound, which was then filtered, washed with hexane and dried under reduced pressure at 50 C. The yield was 1.30 g (86.89%); HPLC purity-99.8% |
| 90% |
With hydrogenchloride; In water; at 2 - 70℃; for 20h; |
Example 5 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxy-camptothecin hydrochloride Trihydrate (CPT-11) In a suitable vessel were charged 7-ethyl-10-[4-(1-piperidino)-1-piperidino] carbonyloxy-camptothecin (27gm) obtained in Example 2 and purified water. Subsequently, concentrated hydrochloric acid (5.76 gm) was added to dissolve the reaction mass completely. Reaction mass was treated with activated carbon and filtered. Further concentrated hydrochloric acid (13.5 gm) was added to the filtrate at 60-70 C and the reaction mixture was maintained at 2-5 C for 20 hours. The product so obtained was filtered, washed with isopropanol and dried under reduced pressure at 50C. The yield was 28 g (90%); HPLC purity 100%. i) a powder X-ray diffraction pattern substantially in accordance with Figure 1; ii) a powder X-ray diffraction pattern having peaks at about 7.60, 8.21, 9.55, 10.96, 12.34, 14.33, 15.79, 19.92, 21.25, 22.73, 24.79, 25.99 and 27.68 +/- 0.2 degrees 20; |
| 90% |
With hydrogenchloride; In water; at 70℃; for 3h; |
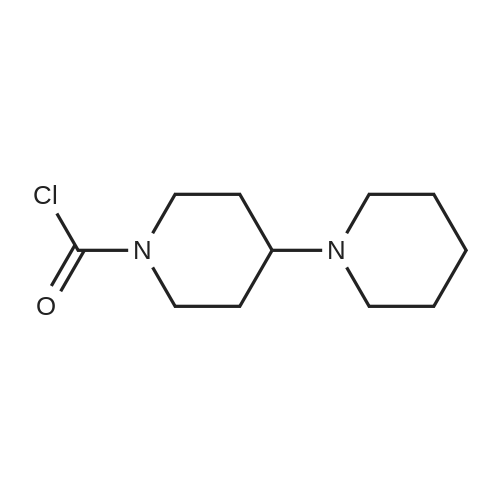
Example 4: Irinotecan hydrochloride7-Ethyl-lO-hydroxycamptothecin (20 g) was suspended in methylene chloride (400 ml). To this while stirring at room temperature l-chlorocarbonyl-4-piperidinopiperidine hydrochloride (27.2 g; 2 eq.) and N-methylpyrrolidine (40.0 ml; 7 eq.) was added. There was visible temperature raise of 5C in the next 10 to 20 minutes and stirred for further 30 minutes to dissolve, all the suspended material into solution. The clear solution was further stirred for additional 2 hours (In process check shows the absence of SN-38). The solvent was removed along with excess of N-Methylpyrrolidine under reduced pressure, keeping the temperature below 45C. After cooling the solid mass was treated with water (250 ml) and stirred. To this aqueous solution, methylene chloride (1 L) was added and stirred well to extract all the free base of irinotecan. The organic layer was collected, washed with water twice (250 ml x 2), solvent was removed to give pale yellowish solid The free base is suspended in 280 ml of water and to this 16.3 ml of concentrated hydrochloride (3 eq.) was added and stirred at room temperature for 15 minutes to form clear solution. The solution was then heated to 70C for 3 hrs and slowly allowed to cool to room temperature. It was further cooled to 0-5C for 30 minutes, collected the solid, washed with water (60 ml), ethanol (60 ml) and dried at room temperature to give 30 g of irinotecan hydrochloride, purity 99.5% (yield 90%).The above compound is further purified by taking in isopropyl alcohol -water mixture (24 ml); (6 ml isopropyl alcohol and 18 ml of H20) and heated to 70C and adjusted the pH to 3.5 to 3.8 by adding 5% hydrochloride (0.2 ml) to form clear solution. The solution was then allowed to cool to room temperature, collected the product by filtration, washed with IPA-H20 (1 :3) and dried at room temperature to give 2.6 g of colourless crystalline compound of irinotecan hydrochloride of 99.78% purity by HPLC with no impurity >0.1%. |
| 89.8% |
|
SN-38B-11 (1.00 g, 1.7 mmol) obtained in Example 10 was suspended in 1/10 N hydrochloric acid (20 mL, 2.0 mmol), and the suspension was heated at about 80C to dissolve in. Acetonitrile (100 mL) was added to the solution, and the mixture was stirred at room temperature for overnight. The precipitates were filtered, dried, and humidified under 75% RH afforded CPT-11 (0.95 mg, yield 89.8%) as pale yellow crystalline powder. |
| 85.6% |
With hydrogenchloride; In tetrahydrofuran; water;Medicinal carbon present;Product distribution / selectivity; |
One gram of irinotecan was added to 120 mL of tetrahydrofuran. The resultant solution was stirred for 30 minutes in the presence or absence of 0.2 g of medicinal carbon. The medicinal carbon was filtered off by a membrane filter. While stirring the filtrate, 6 mol/L hydrochloric acid was added in an amount 1.05 times by mole as large as irinotecan and subjected to the same process as in Example 1. Note that the moisture absorbing time until the solution reached constant weight was about 100 hours. The results of infrared absorption spectrum and thermoanalysis (differential scanning calorimetry) for crystals obtained ina systemhavingmedicinal carbon added thereto are shown in Figures 4 and 5. It was confirmed that the obtained crystals is c-type crystals. The yield of the c-type crystals was 85.6% in the case where medicinal carbon was added and 100% in the case where no medicinal carbon was added. |
| 76.3 - 83.4% |
With hydrogenchloride; In water; acetonitrile; at 22℃; for 25 - 46h;Product distribution / selectivity; |
Irinotecan was suspended in acetonitrile or acetone in accordance with the amounts shown in Table 1 and dissolved by adding, 6 mol/L hydrochloric acid. To each mixture solution, 1 mg of c-type crystals separately prepared was added and the solution was stirred at 22C for 25 to 46 hours. The formed crystals were obtained by filtration, dried under reduced pressure, and subjected to a moisture absorption process performed by a saturated aqueous sodium chloride solution method until the crystals showed constant weight (about 80 hours). In this manner, the crystals of irinotecan hydrochloride were obtained. Table 1 shows the results of the preparations. [Table 1] No.Irinotecan (g)Solvent1) Hydrochloric acid2) Seed crystalsCrystallization time (hour)Yield (%)AcetonitrileAcetone 15.0801.0-4683.425.01201.0c form2580.5310.01201.05c form4176.341.03601.05c form4191.71) mL/g (Irinotecan)2) Times by mole relative to irinotecan The crystals of irinotecan hydrochloride obtained above were subjected to infrared absorption spectrum analysis, powder X-ray diffraction analysis, and thermoanalysis. The results are shown in Figures 1 to 3. In the infrared absorption spectrum, strong absorption was observed at wavelengths of about 1757 cm-1, 1712 cm-1, 1667 cm-1 (Figure 1). In the powder X-ray diffraction, diffraction peaks (2theta) were observed at 9.15, 10.00, 11.80, 12.20, 13.00 and 13.40; however no strong peak was observed at 11.00, which is intrinsic to the b-type crystals (Figure 2). In the thermoanalysis (differential scanning calorimetry), an endothermic peak near 90C due to dehydration was not observed, which is intrinsically observed in the b-type crystals (Figure 3). The moisture content of the crystals was measured by the Karl Fischer method. As a result, the moisture content of No.3 was 3.96%. Therefore, it turned out that the crystals above were obtained in the form of sesquihydrates (calculation value: 4.15%). From the results above, it was confirmed that the crystals of irinotecan hydrochloride prepared in accordance with Nos. 1 to 4 are c-type, which are different from the b-type crystals conventionally crystallized from water. |
|
With hydrogenchloride; In water; at 60℃; |
A solution of 4-amino-3-propionylphenyl-1,4'-bipiperidine-1'-carboxylate as prepared in Example 1 (0.359 g, 1.0 mmol) and 14CPT (the compound of formula (I) above) (0.39 g, 1.5 mmol) in 10 ml acetic acid was heated to 115 C. for 22 hours. The resulting mixture was distilled under vacuum to yield a black semisolid residue. The residue was dissolved in a 95:5 (v/v) methylene chloride/methanol mixture and chromatographed on 20 g silica-60 (230-400 mesh), eluting with 95:5 (v/v) methylene chloride/methanol (400 ml) and then 92.5:7.5 (v/v) methylene chloride/methanol (600 ml). The resulting product fractions were combined and evaporated to provide a residue, which was then redissolved in 20 ml methylene chloride and washed with saturated aqueous sodium bicarbonate solution (2×10 ml). The resulting organic phase was dried over sodium sulfate and evaporated. Ethanol was added and the resulting solution concentrated to a volume of 8 ml before being allowed to crystallize overnight. The solids were filtered and dried to yield 0.243 g of irinotecan (CPT-11 free base) as a pale yellow solid. The irinotecan produced as above (0.216 g) was dissolved in 2 ml water and 0.43 ml 1M HCl and heated to 60 C. to form a yellow solution. This solution was filtered hot over powdered activated carbon (Darco m G-60) (0.5 g). The filtrate was cooled and seeded with 5 mg irinotecan hydrochloride crystals and allowed to crystallize overnight. The solids were filtered, washed with water (2×1 ml), and suction dried under air to yield 0.123 g of irinotecan hydrochloride trihydrate as pale yellow crystals having a melting point of 259.8 C. |
|
With hydrogenchloride; In water; at 20℃; for 50h;Product distribution / selectivity; |
Ten grams of irinotecan was suspended in about 100 mL of purified water. About 10 mL of diluted hydrochloric acid (the Japanese Pharmacopoeia) was added dropwise to the suspension solution while stirring. Then, irinotecan was dissolved by raising temperature of the suspension solution. After the resultant solution was filtered, a seed crystals was added to the filtrate, whichwas stirred at room temperature for about 50 hours to form crystals. The resultant crystals were subjected to suction filtration and a filtrated product was dried under reduced pressure. A dried product was allowed to stand still in a humidifier to humidify in accordance with a saturated aqueous sodium chloride solution method. |
|
With hydrogenchloride; In water; at 60℃; |
A solution of 4-amino-3-propionylphenyl-1, 4';-bipiperidine-1';-carboxylate as prepared in Example 1 (0.359 g, 1.0 mmol) and 14CPT (the compound of formula (I) above) (0. 39 g, 1.5 mmol) in 10 rnl acetic acid was heated to 115C for 22 hours. The resulting mixture was distilled under vacuum to yield a black semisolid residue. The residue was dissolved in a 95: 5 (v/v) methylene chloride/methanol mixture and chromatographed on 20 g silica-60 (230-400 mesh), eluting with 95: 5 (v/v) methylene chloride/methanol (400 ml) and then 92.5 : 7.5 (v/v) methylene chloride/methanol (600 ml). The resulting product fractions were combined and evaporated to provide a residue, which was then redissolved in 20 ml methylene chloride and washed with saturated aqueous sodium bicarbonate solution (2 X 10 ml). The resulting organic phase was dried over sodium sulfate and evaporated. Ethanol was added and the resulting solution concentrated to a volume of 8 rnl before being allowed to crystallize overnight. The solids were filtered and dried to yield 0.243 g of irinotecan (CPT-11 free base) as a pale yellow solid. The irinotecan produced as above (0.216 g) was dissolved in 2 ml water and 0.43 ml 1M HC1 and heated to 60 C to form a yellow solution. This solution was filtered hot over powdered activated carbon (Darco G-60) (0.5 g). The filtrate was cooled and seeded with 5 mg irinotecan hydrochloride crystals and allowed to crystallize overnight. The solids were filtered, washed with water (2 x 1 ml), and suction dried under air to yield 0.123 g of irinotecan hydrochloride trihydrate as pale yellow crystals having a melting point of 259. 8C. |
|
With hydrogenchloride; In water; at 70℃; for 3h;Product distribution / selectivity; |
EXAMPLE 10 Preparation of Irinotecan Hydrochloride Trihydrate 25 g of irinotecan prepared according to the procedure of Example 9 was slurried with 250 ml of water for about 30 minutes and filtered, the solid was washed with 12.5 ml of water, 27.5 ml of 5N hydrochloric acid was added to the filtrate, and the filtrate was heated to 70 C. with simultaneous stirring for 3 hours at 70 C. until complete precipitation occurred. The reaction mass was cooled to 30 C. and filtered, followed by washing the solid with 7 ml of water and 15 ml ethanol. Finally the solid was dried under a vacuum of about 650 mm Hg at 30 C. for 3 hours to afford 22.5 g of the title compound with a purity by HPLC of 99.95%. EXAMPLE 11; Preparation of Irinotecan Hydrochloride Trihydrate; 290 g of irinotecan prepared according to the procedure of Example 9 was slurried with 2.9 L of water and 87 ml of 5N hydrochloric acid was added over about 30 minutes. The mixture was filtered through a 0.22 micron filter, and then 232 ml of 5N hydrochloric acid was added to the filtrate. The filtrate was heated to 70 C. with simultaneous stirring and stirred for about 3 hours at 70 C. until precipitation occurred. The reaction mass was cooled to 30 C. and filtered, followed by washing the solid with 87 ml of water and 0.174 L of ethanol. Finally the solid was dried under a vacuum of not less than 580 mm Hg at 30 C. for about 3 hours to afford 257 g of the title compound. |
|
With hydrogenchloride; In water; at 60℃; for 0.5h;Product distribution / selectivity; |
Step IV: Preparation oflrinotecan Hydrochloride TrihydrateTake Step- III (10.4g) product in DM H2O (93.6ml) and add cone. HCl (19.80ml). Heat the mixture to 60when a pale yellow solution is obtained. Add activated charcoal (1.04g) and continue to maintain the mixture at this temperature for further 30 min and filter. Cool the filtered solution to room temperature and then to 0C with stirring for an overnight. Filter the separated solid, dry the solid under high vacuum to yield irinotecan hydrochloride trihydrate (6.13g); HPLC purity 99.65%; Step IV: Preparation oflrinotecan Hydrochloride Trihydrate: Take Step- III (10.Og) product in DM H2O (90.0ml) and add cone. HCl (19.00ml). Heat the mixture to 60when a pale yellow solution is obtained. Add activated charcoal (1.0Og) and continue to maintain the mixture at this temperature for further 30 min and filter.Cool the filtered solution to room temperature and then to 00C with stirring for an overnight. Filter the separated solid, dry the solid under high vacuum to yield irinotecan hydrochloride trihydrate (5.9g); HPLC purity 99.60%; Step IV: Preparation of Irinotecan Hydrochloride TrihydrateTake Step- III (10.0 g) product in DM H2O (90.0ml) and add cone. HCl (19. OmI). Heat the mixture to 60when a pale yellow solution is obtained. Add activated charcoal (1.04g) and continue to maintain the mixture at this temperature for further 30 min and filter.Cool the filtered solution to room temperature and then to 00C with stirring for an overnight. Filter the separated solid, dry the solid under high vacuum to yield irinotecan hydrochloride trihydrate (6.03g); HPLC purity 99.62% |
|
With hydrogenchloride; In methanol; water; at 50℃; |
Example 2 - Preparation of irinotecan form H from irinotecan base A mixture of irinotecan free base (10.0 g) in methanol (500 ml) was heated to 50C. 37% hydrochloric acid in water (1.4 ml) was dripped into the solution, and a solution was obtained. The solvent was distilled at a pressure of under 50 mbars at 50C until a dry residue was obtained. The concentrated mass was taken up with dichloromethane (150 ml) and left under stirring at 35C until a solution was obtained. The solvent was distilled at a pressure of under 300 mbars at 35C, until a concentrated solution (100 ml) was obtained, which was left to cool to 25C; the formation of the first crystals of the product was observed after approx. 30 minutes. After 15 hours' curing at 25C, the crystallised mass was filtered through a Buchner funnel under suction, and the wet product was washed with dichloromethane (35 ml). After oven-drying under vacuum (residual pressure under 50 mbars), irinotecan hydrochloride form H (8.2 g) with high purity (HPLC purity = 99.6%) and a 4.6% water content was obtained. Said product (5.0 g) was hydrated by exposure in an atmosphere with 75% relative humidity. After 18 hours the weight of the product proved stable (5.3 g). Irinotecan hydrochloride form H with high purity was obtained (HPLC purity = 99.6%; water = 9%). |
|
With hydrogenchloride; In water; at 50℃; |
A mixture of irinotecan free base (10.0 g) in methanol (500 ml) was heated to 50 C. 37% hydrochloric acid in water (1.4 ml) was dripped into the solution, and a solution was obtained. The solvent was distilled at a pressure of under 50 mbars at 50 C. until a dry residue was obtained. The concentrated mass was taken up with dichloromethane (150 ml) and left under stirring at 35 C. until a solution was obtained. The solvent was distilled at a pressure of under 300 mbars at 35 C., until a concentrated solution (100 ml) was obtained, which was left to cool to 25 C.; the formation of the first crystals of the product was observed after approx. 30 minutes. After 15 hours' curing at 25 C., the crystallised mass was filtered through a Buchner funnel under suction, and the wet product was washed with dichloromethane (35 ml). After oven-drying under vacuum (residual pressure under 50 mbars), irinotecan hydrochloride form H (8.2 g) with high purity (HPLC purity=99.6%) and a 4.6% water content was obtained.Said product (5.0 g) was hydrated by exposure in an atmosphere with 75% relative humidity. After 18 hours the weight of the product proved stable (5.3 g). Irinotecan hydrochloride form H with high purity was obtained (HPLC purity=99.6%; water=9%). |
|
With hydrogenchloride; In water; for 1 - 4h;Product distribution / selectivity; |
Example-6; Preparation of Irinotecan hydrochloride trihydrate (IRT.HCI.3H20); Charging IRT-5 (30g) obtained in example 5 onto a mixture of DM water (1300 ml) and concentrated hydrochloride acid (28 ml), stirring for 1 to 4 hour to dissolve completely, washing the solution thus obtained with chloroform, collecting aqueous solution. Treating the aqueous solution with carbon under stirring, filtering, collecting filtrate and distilling partially water from washed filtrate under vacuum at a temperature below 45C, cooling to 0 to 5C for a period of 10 hours to 12 hours, filter the solid obtained, drying under vacuum below 45C to obtain Irinotecan hydrochloride trihydrate (22.5g). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping