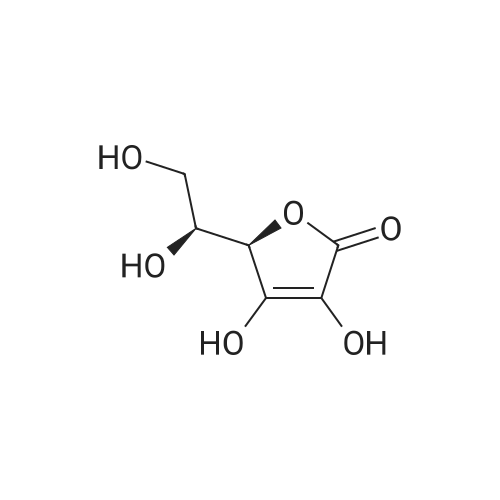
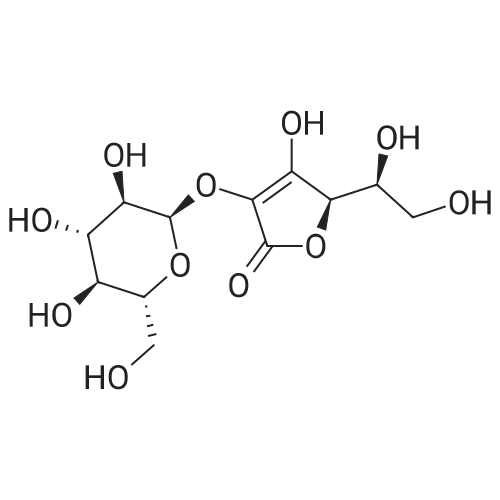
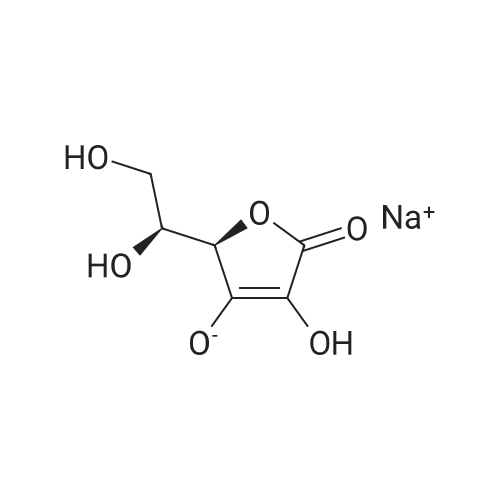
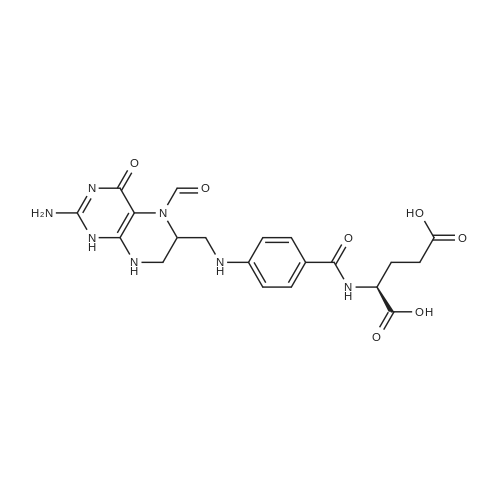
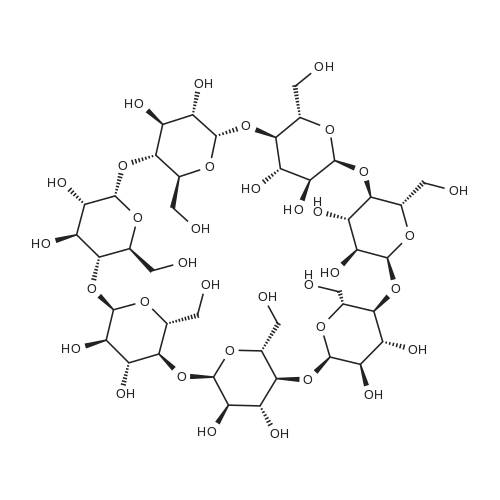
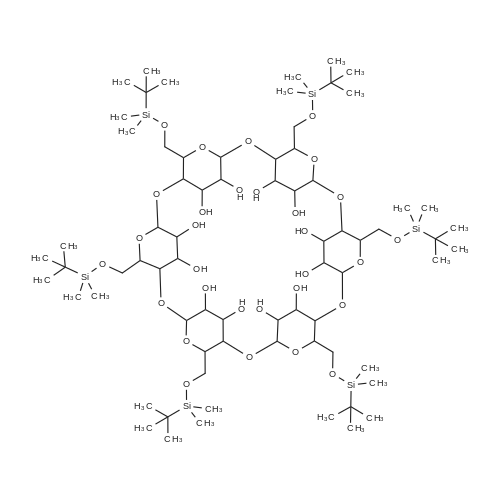
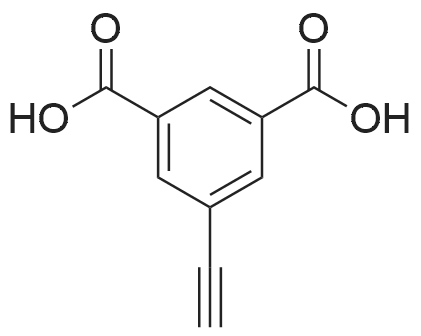
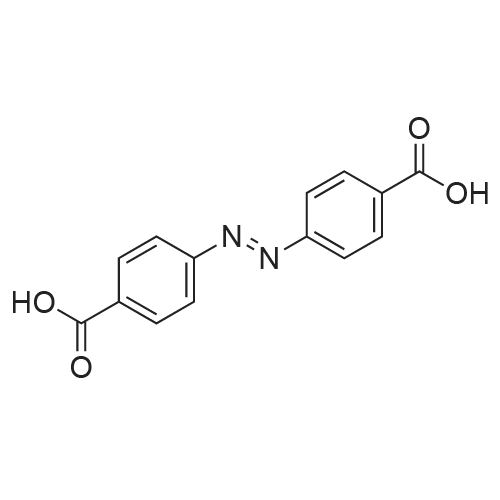
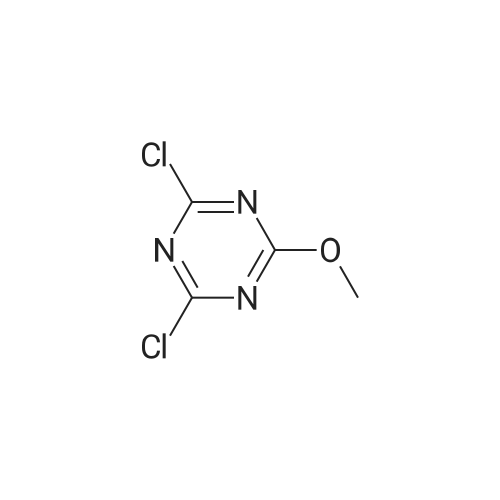
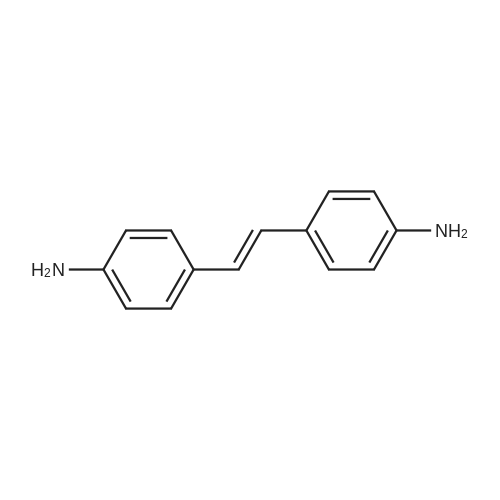
| 27% |
Stage #1: alpha cyclodextrin With lithium hydride In dimethyl sulfoxide for 24h; Inert atmosphere;
Stage #2: allyl bromide With lithium iodide In dimethyl sulfoxide for 2h; Inert atmosphere;
Stage #3: acetic anhydride With triethylamine at 80℃; |
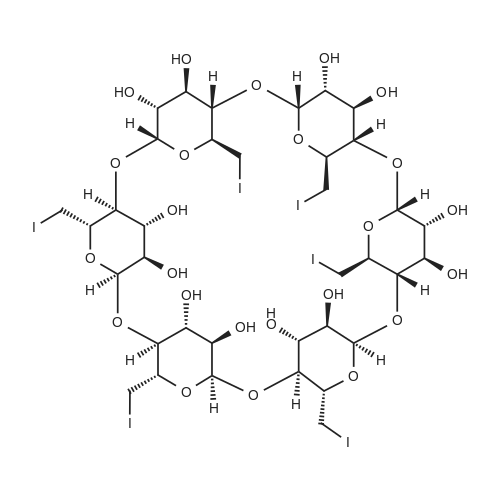
4.2.1. Per-O-acetyl-2I-O-allyl-α-cyclodextrin (3a)
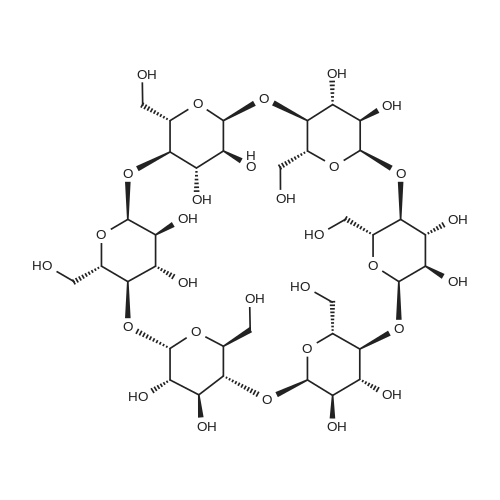
Firstly, the synthesis was carried out according to the exactly the same reaction conditions as described by Hanessian et al.refPreviewPlaceHolder10 The results were the same as described below, but the procedure was inconvenient. Thus the procedure was improved as follows: Dried α-cyclodextrin (880 mg, 910 μmol) was dissolved in dry DMSO (9 mL), and lithium hydride was added (11 mg, 1.4 mmol). The reaction mixture was stirred under an Ar atmosphere for 24 h. Then allyl bromide (79 μL, 910 μmol) and lithium iodide (3 mg, 24 μmol) were added to the reaction mixture. The reaction was monitored by TLC (6:3:1:1 1-PrOH-H2O-EtOAc-aq NH3) and was determined finished after 2 h when no significant increase in monosubstituted derivatives was observed. The CD derivatives were precipitated with acetone (170 mL), filtered out, and washed with acetone (50 mL). The precipitate was adsorbed on silica gel (3.5 g), and mono-O-allyl-α-CDs were separated by chromatography on a silica gel column (7:3:2 1-PrOH-H2O-EtOAc-aq NH3). The mixture of mono-O-allyl-α-CDs was then peracetylated. A suspension of mono-O-allyl-α-CDs in Ac2O (2.2 mL, 24 mmol) and Et3N (2.2 mL, 16 mmol) was stirred at 80 °C overnight. The reaction mixture was diluted with CHCl3, washed with 5% HCl, and the organic layer was evaporated in vacuo to give a brown residue that was purified by chromatography on silica gel (100:1 CHCl3-MeOH). Workup afforded 420 mg (27% overall yield based on α-CD) of the title compound as a white powder: mp 139-141 °C. +98 (CHCl3). IR (KBr): ν = 1747, 1372, 1238, 1040 cm-1. 1H NMR (300 MHz, CDCl3): δ 5.81 (ddt, 1H, J2',3'a 17.1, J2',3'b 10.4, J2',1' 5.7 Hz, H-2'), 5.73 (dd, 1H, J2,3 10.2, J3,4 8.9 Hz, H-3), 5.64 (dd, 1H, J2,3 10.6, J3,4 9.0 Hz, H-3), 5.59 (dd, 1H, J2,3 10.3, J3,4 8.7 Hz, H-3), 5.50 (dd, 1H, J2,3 10.1, J3,4 8.7 Hz, H-3), 5.46 (dd, 1H, J2,3 10.2, J3,4 8.7 Hz, H-3), 5.32 (dd, 1H, J2,3 9.8, J3,4 9.1 Hz, H-3I), 5.24 (dq, 1H, J2',3'a 17.2, J1',3'a = J3'a,3'b 1.5 Hz, H-3'a), 5.19 (dq, 1H, J2',3'b 10.3, J1',3'b = Jgem 1.2 Hz, H-3'b), 5.14 (d, 1H, J1,2 3.5 Hz, H-1), 5.11 (d, 1H, J1,2 3.6 Hz, H-1), 5.09 (d, 1H, J1,2 3.8 Hz, H-1), 4.99 (d, 1H, J1,2 2.9 Hz, H-1), 4.99 (d, 1H, J1,2 3.1 Hz, H-1), 4.87 (d, 1H, J1,2 3.2 Hz, H-1I), 4.82 (dd, 1H, J2,3 10.3, J1,2 3.9 Hz, H-2), 4.81 (dd, 1H, J2,3 10.7, J1,2 3.7 Hz, H-2), 4.77 (dd, 1H, J2,3 10.4, J1,2 3.6 Hz, H-2), 4.77 (dd, 1H, J2,3 10.2, J1,2 3.5 Hz, H-2), 4.71 (dd, 1H, J2,3 10.2, J1,2 3.8 Hz, H-2), 4.60-4.05 (m, 18H, 6 × H-5, 12 × H-6), 4.03-3.99 (m, 2H, 2 × H-1'), 3.85-3.77 (m, 4H, 4 × H-4), 3.74 (t, 1H, J3,4 = J4,5 9.2 Hz, H-4), 3.66 (t, 1H, J3,4 = J4,5 9.2 Hz, H-4I), 3.32 (dd, 1H, J2,3 10.1, J2,3 3.1 Hz, H-2I), 2.30-1.98 (m, 51H, 17 × CH3). 13C NMR (101 MHz, CDCl3): δ 170.90-169.01 (17 × CO), 134.46 (C-2'), 117.76 (C-3'), 98.68 (C-1I), 97.19 (C-1), 96.70 (C-1), 96.58 (C-1), 96.38 (C-1), 95.82 (C-1), 79.58-68.73 (5 × C-2, 5 × C-3, 5 × C-4, 6 × C-5), 78.06 (C-4I), 77.96 (C-2I), 72.87 (C-3I), 72.13 (C-1'), 63.40-62.89 (6 × C-6), 20.94-20.62 (17 × CH3). HRESIMS: [M+Na]+, found 1749.5146. C73H98O47Na requires 1749.5171. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping